Reacción sigmatrópica – Qué es, mecanismo y ejemplos (Cope, Claisen)
Reacción sigmatrópica: explica qué es, mecanismo y ejemplos clásicos Cope y Claisen, reordenamientos [3,3] y aplicaciones en síntesis orgánica
Una reacción sigmatrópica en química orgánica es una reacción pericíclica concertada e intramolecular. En su forma típica no utiliza un catalizador y acontece dentro de una sola molécula: se rompe un enlace σ y, simultáneamente, se forma otro enlace σ distinto. El término sigmatrópico procede de la combinación de «sigma», usado históricamente para los enlaces sencillos carbono‑carbono, y la palabra griega tropos (giro), lo que refleja el desplazamiento del enlace σ durante la reacción.
Se trata de una reordenación intramolecular en la que un sustituyente se desplaza de una región de un sistema π a otra, con reordenación simultánea del sistema π circundante. Como reordenamiento, no hay pérdida ni ganancia neta de átomos: cambia únicamente la conectividad y la posición de los vínculos. Aunque las reacciones sigmatrópicas “clásicas” transcurren sin catalizador, algunas variantes admiten ácidos de Lewis o bases que aceleran el proceso; además, en síntesis moderna existen procedimientos análogos que emplean catalizadores de metales de transición.
Notación y mecanismo. Las sigmatrópicas se clasifican por la notación [i,j], donde i y j indican el número de átomos contados a través de cada rama del sistema π desde el átomo que pierde el enlace σ hasta el átomo que lo gana. Por ejemplo, un reordenamiento [3,3] implica un tránsito a través de un estado de transición cíclico de seis átomos. El mecanismo es concertado y pericíclico: la ruptura y formación de enlaces se producen de manera simultánea a través de un estado de transición con carácter aromático (resonancia estabilizadora), lo que explica la baja entropía de activación y la alta selectividad estereoespecífica de estas reacciones.
Selección orbital y esterequímica. Las reglas de Woodward–Hoffmann describen si una vía suprafacial (el grupo migrante se conecta a la misma cara del sistema π) o antarafacial (cambia de cara) es termodinámica y estereoelectrónicamente permitida bajo condiciones térmicas o fotolíticas. En la práctica, muchos reordenamientos [3,3] (por ejemplo, Cope y Claisen) son térmicamente permitidos por una vía suprafacial–suprafacial que pasa por un estado de transición de seis miembros con geometría tipo silla o bote, lo que conduce a resultados estereoespecíficos y predecibles según la conformación de ese estado de transición.
Tipos y variantes importantes:
- Reordenamiento de Cope (reordenamiento [3,3] de 1,5‑dienos): un ejemplo clásico es la isomerización concertada de un 1,5-dieno a otro 1,5-dieno mediante un estado de transición de seis miembros. Puede ser degenerado (producto igual al reactivo) o no degenerado si hay sustituyentes que estabilizan un isómero sobre otro. La conformación de silla del estado de transición determina la estereoquímica del producto.
- Oxy‑Cope: variante del Cope en que hay un grupo hidroxilo que, tras el reordenamiento [3,3], da lugar a un enol que tautomeriza a una cetona o aldehído. La versión aniónica (anionic oxy‑Cope), en la que la OH se desprotona con base fuerte, acelera enormemente la reacción y la hace prácticamente irreversible hacia la formación de carbonilos.
- Reordenamiento de Claisen (también [3,3]): afecta a éteres vinílicos o arílicos alílicos (por ejemplo, allyl vinyl ethers). El resultado típico es la conversión a compuestos γ,δ‑insaturados o, en el caso de allyl aryl ethers, a fenoles orto‑alquilados (Claisen orto‑rearrangement). Muchas variantes (Ireland–Claisen, Johnson–Claisen, Eschenmoser–Claisen) emplean condiciones o intermedios distintos para controlar la quimio‑ y estereoselectividad.
- Reordenamiento de Carroll y otras reacciones sigmatrópicas especializadas: hay numerosas variantes que amplían la utilidad sintética de estos reordenamientos en la construcción de esqueletos carbonados complejos y en síntesis de productos naturales.
Ejemplos prácticos y utilidad sintética:
- Claisen clásico: un allyl vinyl ether bajo calentamiento sufre un reordenamiento [3,3] dando un enol que luego tautomeriza a una aldehído o cetona γ,δ‑insaturada. En síntesis orgánica, el reordenamiento de Claisen es muy usado para introducir funciones carbonílicas en posiciones remotas y para construir anillos.
- Cope de 1,5‑dienos: la isomerización puede permitir desplazar dobles enlaces, reorganizar sustituyentes y facilitar ciclizaciones posteriores. El oxy‑Cope es especialmente útil para transformar alcoholes en carbonilos y así reorganizar el esqueleto con formación de nuevas funciones.
- Claisen orto: por ejemplo, la conversión de un allyl phenyl ether en un o‑allylphenol es una herramienta clásica para funcionalizar anillos aromáticos de forma regioselectiva.
Condiciones y catalizadores. Aunque muchas sigmatrópicas son térmicas y no requieren catalizador, existen excepciones y variantes catalizadas. Ácidos de Lewis (p. ej., ZnCl2, AlCl3 en algunos casos) o bases fuertes (anionic oxy‑Cope) pueden acelerar la reacción o hacerla irreversible. Además, catalizadores de metales de transición pueden mediar procesos análogos mediante formación de complejos intermedios que recrean la reordenación de enlaces en pasos no estrictamente concertados pero útiles sintéticamente.
Consideraciones prácticas: la velocidad y la dirección del equilibrio dependen de la estabilización relativa de reactivos y productos (sustituyentes conjugantes, estabilización por aromatización parcial o formación de carbonilos en oxy‑Cope), de la conformación que adopta el estado de transición (silla vs. bote) y de la posibilidad geométrica de realizar un camino antarafacial (a menudo estéricamente prohibido en cadenas cortas). Por ello, el diseño de sustratos que favorezcan la conformación de transición adecuada es clave para obtener buenos rendimientos y controlar la estereoquímica.
En resumen, las reacciones sigmatrópicas son reordenamientos pericíclicos concertados que permiten desplazar enlaces σ dentro de una molécula, con gran utilidad en síntesis orgánica para reorganizar esqueletos carbonados y construir nuevas funciones con alta estereospecificidad. Entre las más empleadas en síntesis están el reordenamiento [3,3] de Cope y el reordenamiento de Claisen, junto a sus múltiples variantes (oxy‑Cope, Ireland–Claisen, etc.) que amplían su aplicabilidad en la construcción de moléculas complejas.
Galería de imágenes
10 Imágenes









Visión general de los desplazamientos sigmatrópicos
Nomenclatura del cambio sigmatrópico de Woodward-Hoffman
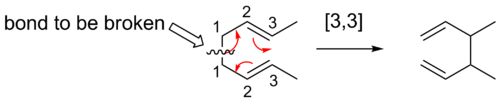
Para describir los desplazamientos sigmatrópicos se utiliza una notación especial. A cada uno de los átomos de carbono de la columna vertebral de la molécula se le asigna un número de posición. Los reordenamientos sigmatrópicos se describen mediante un término de orden [i,j]. Esto significa la migración de un enlace σ adyacente a uno o más sistemas π a una nueva posición (i-1) y (j-1) átomos alejados de la ubicación original del enlace σ. Cuando la suma de i y j es un número par, esto es una indicación de la participación de una cadena de átomos neutros, todo C. Un número impar sugiere que hay un átomo de C cargado o de un par solitario de heteroátomos que sustituye a un doble enlace carbono-carbono. Así, los desplazamientos [1,5] y [3,3] se convierten en [1,4] y [2,3] con heteroátomos, conservando las consideraciones de simetría. Los hidrógenos se omiten en el tercer ejemplo para mayor claridad.

He aquí una manera de encontrar el orden de un reordenamiento sigmatrópico dado. El primer paso es dar números a cada átomo, empezando por los átomos del enlace que se rompe como átomo 1. Los químicos cuentan los átomos en cada dirección desde el enlace roto hasta los átomos que forman el nuevo enlace σ en el producto. Los números que corresponden a los átomos que forman el nuevo enlace se separan con una coma y se colocan entre paréntesis. Así se crea el descriptor de orden de reacción sigmatrópico.
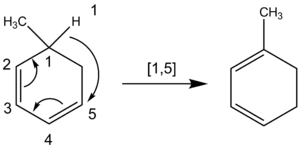
Los químicos también cuentan los átomos cuando nombran un desplazamiento sigmatrópico en el que se desplaza un átomo de hidrógeno. La cadena de carbono no se rompe en una migración de un átomo de hidrógeno. Por ello, los químicos cuentan a través de todos los átomos implicados en la reacción en lugar de sólo a través de los átomos más cercanos. Por ejemplo, la siguiente migración de átomos de hidrógeno es del orden [1,5], que se obtiene contando en sentido contrario a las agujas del reloj a través del sistema π, en lugar del orden [1,3] a través del grupo CH del anillo 2que resultaría erróneamente si se contara en el sentido de las agujas del reloj.
Desplazamientos suprafaciales y antarafaciales
Los químicos han estudiado las reacciones sigmatrópicas en las que el grupo que migra tiene un esterocentro. En principio, todos los desplazamientos sigmatrópicos pueden producirse con la misma geometría (retención) o con la opuesta (inversión) del grupo que migra. Esto depende de si se utiliza el lóbulo de enlace original del átomo que migra o su otro lóbulo para formar el nuevo enlace.
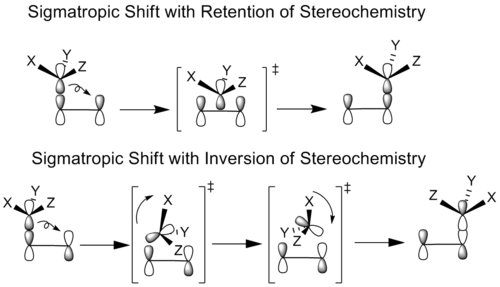
En los casos de retención estereoquímica, el grupo migratorio se traslada sin rotación a la posición de enlace. En el caso de la inversión estereoquímica, el grupo migratorio rota y se traslada para alcanzar su conformación de enlace.
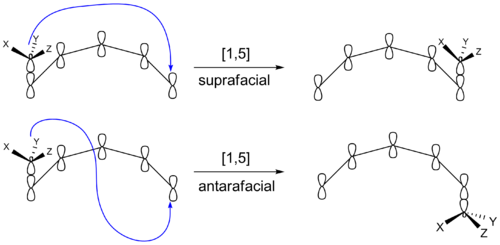
Hay otra forma en la que una reacción sigmatrópica puede producir productos con diferente esteroquímica. El grupo que migra puede permanecer en la cara original del sistema π después de la reconexión. O bien, puede ir a la cara opuesta del sistema π. Si el grupo migrante permanece en la misma cara del sistema π, el cambio se conoce como suprafacial. Si el grupo que migra se traslada a la cara opuesta se denomina desplazamiento antarafacial. Las transformaciones que ocurren dentro de anillos pequeños o medianos no pueden realizar desplazamientos antarafaciales.
Clases de reordenamientos sigmatrópicos
[1,3] Turnos
Desplazamientos térmicos de los hidruros
En un desplazamiento térmico [1,3] del hidruro, éste se desplaza tres átomos. Las reglas de Woodward-Hoffmann dictan que se produciría un desplazamiento antarafacial. Aunque tal desplazamiento está permitido por la simetría, la topología de Mobius requerida en el estado de transición prohíbe tal desplazamiento. Es geométricamente imposible. Por eso los enoles no se isomerizan sin un catalizador ácido o básico.
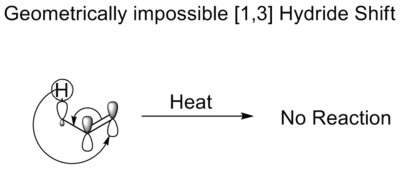
Desplazamientos térmicos de alquilos
Los desplazamientos térmicos del alquilo [1,3], similares a los desplazamientos del hidruro [1,3], deben proceder antarafacialmente. La geometría del estado de transición es prohibitiva. Pero un grupo alquilo, debido a la naturaleza de sus orbitales, puede invertir su geometría y formar un nuevo enlace con el lóbulo posterior de su orbital sp3. Esta reacción dará lugar a un desplazamiento suprafacial. Estas reacciones aún no son comunes en sistemas de cadena abierta debido a la naturaleza altamente ordenada del estado de transición. Por ello, las reacciones funcionan mejor en moléculas cíclicas.
![[1,3] Alkyl Shifts](https://www.alegsaonline.com/image/550px-1%2C3alkylfixed.png)
Cambios fotoquímicos [1,3]
Los desplazamientos fotoquímicos [1,3] deberían ser desplazamientos suprafaciales; sin embargo, la mayoría de ellos no son concertados porque proceden a través de un estado triplete (es decir, tienen un mecanismo diradical, al que no se aplican las reglas de Woodward-Hoffmann).
[1,5] Turnos
Un desplazamiento [1,5] implica el desplazamiento de 1 sustituyente (-H, -R o -Ar) hacia abajo de 5 átomos de un sistema π. Se ha demostrado que el hidrógeno se desplaza tanto en sistemas cíclicos como de cadena abierta a temperaturas iguales o superiores a 200 ˚C. Se predice que estas reacciones proceden de forma suprafacial, mediante un estado de transición de topología Huckel.
![[1,5] Hydride shift in a cyclic system](https://www.alegsaonline.com/image/300px-1%2C5hydridecyclicfixed.png)
La fotoirradiación requeriría un desplazamiento antarafacial del hidrógeno. Aunque estas reacciones son raras, hay ejemplos en los que se favorecen los desplazamientos antarafaciales:
![Antarafacial [1,5] Hydride Shift](https://www.alegsaonline.com/image/600px-1%2C5hantarafacialfixed.png)
A diferencia de los desplazamientos [1,5] del hidrógeno, nunca se han observado desplazamientos [1,5] del alquilo en un sistema de cadena abierta. Los químicos han determinado las preferencias de velocidad para los desplazamientos de alquilo [1,5] en sistemas cíclicos: carbonilo y carboxilo> hidruro> fenilo y vinilo>> alquilo.
Los grupos alquilos sufren desplazamientos [1,5] muy poco y suelen requerir altas temperaturas. Sin embargo, en el caso de los ciclohexadienos, la temperatura para los desplazamientos de los alquilos no es mucho mayor que la de los carbonilos, el mejor grupo migratorio. Un estudio demostró que esto se debe a que los desplazamientos de alquilo en los ciclohexadienos proceden a través de un mecanismo diferente. Primero se abre el anillo, seguido de un desplazamiento [1,7], y luego el anillo se reforma electrocíclicamente:
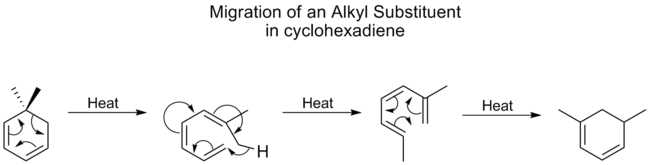
Este mismo proceso mecánico se ve a continuación, sin la reacción final de cierre de anillo electrocíclico, en la interconversión de lumisterol a vitamina D 2.
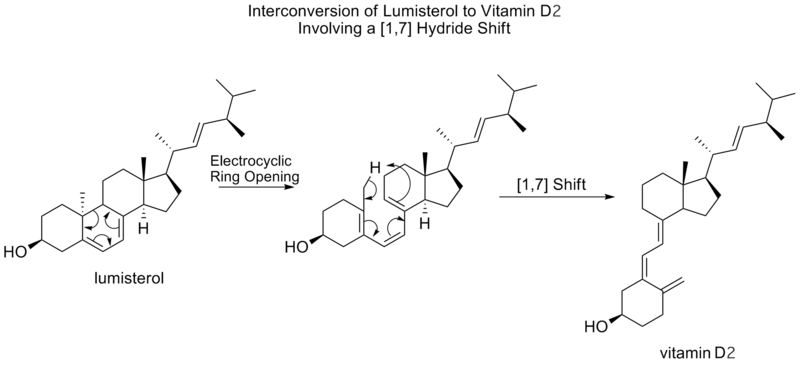
[1,7] Turnos
Los desplazamientos sigmatrópicos [1,7] son predichos por las reglas de Woodward-Hoffmann para proceder de manera antarafacial, por un estado de transición de topología de Mobius. Un desplazamiento antarafacial [1,7] se observa en la conversión de lumisterol a vitamina D 2, donde tras una apertura del anillo electrocíclico a previtamina D 2, se desplaza un hidrógeno de metilo.
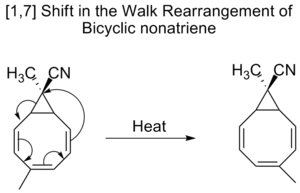
Los nonatrienos bicíclicos también sufren desplazamientos [1,7] en el llamado reordenamiento walk, que es el desplazamiento del grupo divalente, como parte de un anillo de tres miembros, en una molécula bicíclica.
[3,3] Turnos
Los desplazamientos sigmatrópicos [3,3] son reordenamientos sigmatrópicos bien estudiados. Las reglas de Woodward-Hoffman predicen que estas reacciones de seis electrones procederían suprafacialmente, utilizando un estado de transición de topología Huckel.
Reordenamiento de Claisen
Descubierto en 1912 por Rainer Ludwig Claisen, el reordenamiento de Claisen es el primer ejemplo registrado de un reordenamiento [3,3]-sigmatrópico. Este reordenamiento es una útil reacción de formación de enlaces carbono-carbono. Un ejemplo de reordenamiento de Claisen es el reordenamiento [3,3] de un éter vinílico alílico, que al calentarse da lugar a un carbonilo γ,δ-insaturado. La formación de un grupo carbonilo hace que esta reacción, a diferencia de otros reordenamientos sigmatrópicos, sea intrínsecamente irreversible.

Reordenamiento aromático de Claisen
El reordenamiento orto-Claisen implica el desplazamiento [3,3] de un éter fenílico alílico a un intermedio que rápidamente se tautomeriza a un fenol ortosustituido.
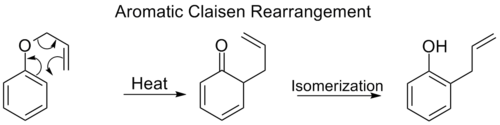
Cuando se bloquean las dos posiciones orto del anillo de benceno, se produce un segundo reordenamiento orto-Claisen. Este reordenamiento para-Claisen termina con la tautomerización a un fenol trisustituido.
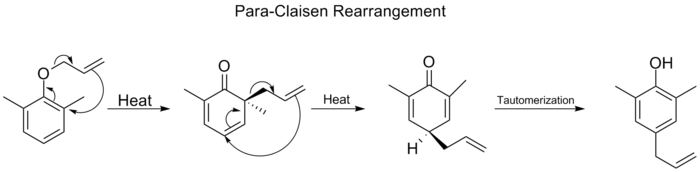
Reordenación de Cope
El reordenamiento de Cope es una reacción orgánica ampliamente estudiada que implica el reordenamiento sigmatrópico [3,3] de 1,5-dienos. Fue desarrollada por Arthur C. Cope. Por ejemplo, el 3,4-dimetil-1,5-hexadieno calentado a 300 °C produce 2,6-octadieno.

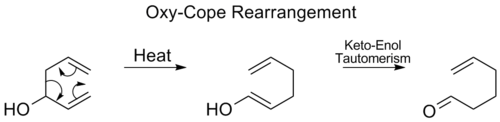
Reordenación de Oxy-Cope
En el reordenamiento Oxy-Cope se añade un grupo hidroxilo en C3 formando un enal o enona tras el tautomerismo Keto-enol del enol intermedio:
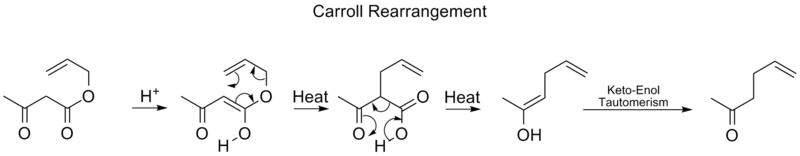
Reordenación de Carroll
El reordenamiento de Carroll es una reacción de reordenamiento en química orgánica y consiste en la transformación de un β-ceto éster alílico en un ácido α-alil-β-cetocarboxílico. Esta reacción orgánica puede ir seguida de una descarboxilación y el producto final es una γ,δ-alilcetona. El reordenamiento de Carroll es una adaptación del reordenamiento de Claisen y efectivamente una alilación descarboxilante.
Síntesis de indoles de Fischer
La síntesis del indol de Fischer es una reacción química que produce el heterociclo aromático indol a partir de una fenilhidrazina (sustituida) y un aldehído o una cetona en condiciones ácidas. La reacción fue descubierta en 1883 por Hermann Emil Fischer.
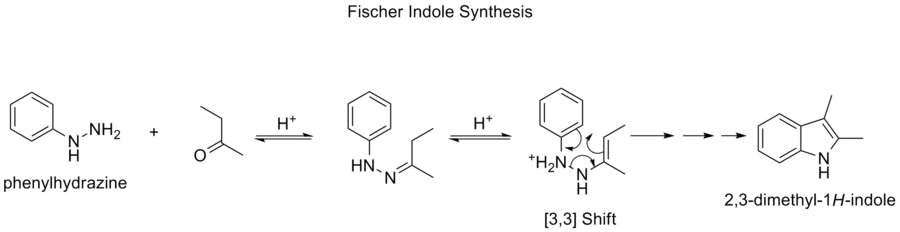
La elección del catalizador ácido es muy importante. Entre los catalizadores ácidos que tienen éxito se encuentran: Los ácidos de Bronsted, como el HCl, el H2SO4 , el ácido polifosfórico y el ácido p-toluenosulfónico. Los ácidos de Lewis, como el trifluoruro de boro, el cloruro de zinc, el cloruro de hierro y el cloruro de aluminio, también son catalizadores útiles.
Se han publicado varias reseñas.
[5,5] Turnos
De forma similar a los desplazamientos [3,3], las reglas de Woodward-Hoffman predicen que los desplazamientos sigmatrópicos [5,5] se producirían de forma suprafacial, estado de transición de la topología de Huckel. Estas reacciones son más raras que los desplazamientos sigmatrópicos [3,3], pero esto es principalmente una función del hecho de que las moléculas que pueden sufrir desplazamientos [5,5] son más raras que las moléculas que pueden sufrir desplazamientos [3,3].
![[5,5] shift of phenyl pentadienyl ether](https://www.alegsaonline.com/image/800px-5%2C5shiftfixeds.png)
Reordenación de la marcha
La migración de un grupo divalente, como el O, el S, el NR o el CR2 , que forma parte de un anillo de tres miembros en una molécula bicíclica, se denomina comúnmente reordenamiento de paseo. Esto puede caracterizarse formalmente según las reglas de Woodward-Hofmann como un desplazamiento sigmatrópico (1, n). Un ejemplo de este tipo de reordenación es el desplazamiento de los sustituyentes en los tropilidenos (1,3,5-cicloheptatrienos). Cuando se calienta, el sistema pi pasa por un cierre de anillo electrocíclico para formar bici[4,1,0]heptadieno (norcaradieno). A continuación se produce un desplazamiento de alquilo [1,5] y una apertura de anillo electrocíclica.
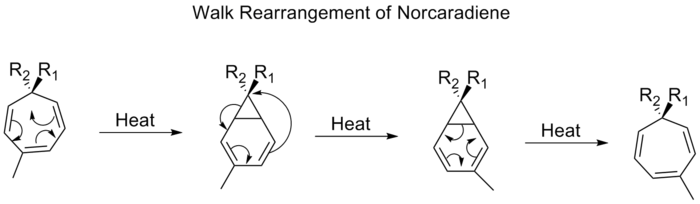
Procediendo a través de un desplazamiento [1,5], se espera que el reordenamiento de los norcaradienos proceda suprafacialmente con una retención de la estereoquímica. Sin embargo, las observaciones experimentales muestran que los desplazamientos 1,5 de los norcaradienos proceden antaracialmente. Los cálculos teóricos encontraron que el desplazamiento [1,5] es un proceso dirádico, pero sin implicar ningún mínimo dirádico en la superficie de energía potencial.
Páginas relacionadas
- Reordenamiento 2,3-sigmotrópico
- Cambio en el NIH
- Teoría de los orbitales moleculares de frontera
- Reglas Woodward-Hoffmann
Preguntas y respuestas
P: ¿Qué es una reacción sigmatrópica en química orgánica?
R: Una reacción sigmatrópica es una reacción pericíclica que implica un proceso intramolecular no catalizado y cambia un enlace σ por otro enlace σ diferente.
P: ¿Una reacción sigmatrópica implica un catalizador?
R: Una reacción sigmatrópica real no suele implicar un catalizador, aunque algunas reacciones sigmatrópicas pueden ser catalizadas por un ácido de Lewis.
P: ¿Qué significa el término "sigmatrópico"?
R: El término "sigmatrópico" es una palabra compuesta formada por "sigma", que se refiere a los enlaces simples carbono-carbono, y la palabra griega "tropos", que significa giro.
P: ¿Qué tipo de reacción es una reacción sigmatrópica?
R: Una reacción sigmatrópica es una reacción de reordenación, lo que significa que los enlaces de una molécula se desplazan entre los átomos sin que salga ningún átomo ni se añadan átomos nuevos a la molécula.
P: ¿Qué ocurre en una reacción sigmatrópica intramolecular?
R: En una reacción sigmatrópica intramolecular, un sustituyente se desplaza de una parte de un sistema de enlace π a otra parte con reordenamiento simultáneo del sistema π.
P: ¿Existen reordenamientos sigmatrópicos bien conocidos?
R: Algunos de los reordenamientos sigmatrópicos más conocidos son el reordenamiento [3,3] de Cope, el reordenamiento de Claisen, el reordenamiento de Carroll y la síntesis del indol de Fischer.
P: ¿Las reacciones sigmatrópicas suelen implicar catalizadores de metales de transición?
R: Sí, las reacciones sigmatrópicas suelen tener catalizadores de metales de transición que forman intermedios en reacciones análogas.
Artículos relacionados
Autor
AlegsaOnline.com Reacción sigmatrópica – Qué es, mecanismo y ejemplos (Cope, Claisen) Leandro Alegsa
URL: https://es.alegsaonline.com/art/90314
Fuentes
- dx.doi.org : 10.1021/ja01044a027
- dx.doi.org : 10.1039/C39760000873
- dx.doi.org : 10.1021/ja01034a076
- dx.doi.org : 10.1016/0040-4039(75)80050-5
- dx.doi.org : 10.1002/anie.197208321
- dx.doi.org : 10.1002/cber.19120450348
- dx.doi.org : 10.1002/cber.19250580207
- dx.doi.org : 10.1002/cber.19260590927
- dx.doi.org : 10.1021/ja01859a055
- dx.doi.org : 10.1021/ja00754a042
- dx.doi.org : 10.1021/ja00026a007
- dx.doi.org : 10.1021/ja01076a067
- dx.doi.org : 10.1039/JR9400000704
- dx.doi.org : 10.1002/cber.188301602141
- dx.doi.org : 10.1002/cber.188401701155