Enfermedad de Creutzfeldt-Jakob: definición, causas (priones) y síntomas
ECJ (Enfermedad de Creutzfeldt-Jakob): definición, causas por priones, síntomas, evolución rápida y pronóstico fatal. Guía clara y actualizada.
La enfermedad de Creutzfeldt-Jakob o ECJ es una enfermedad neurológica. Es degenerativa (empeora con el tiempo), no se puede curar y siempre causa la muerte. La ECJ se denomina a veces una forma humana de la "enfermedad de las vacas locas" (encefalopatía espongiforme bovina o EEB). La EEB es en realidad la causa de un tipo raro de la enfermedad de Creutzfeldt-Jakob; ambas no son la misma enfermedad.
La ECJ está causada por un agente infeccioso llamado prión. Los priones son proteínas que están mal plegadas. Los priones hacen copias de sí mismos cambiando las proteínas correctamente plegadas a formas mal plegadas. La enfermedad de Creutzfeldt-Jakob hace que el tejido cerebral se vuelva insalubre muy rápidamente. A medida que la enfermedad destruye el cerebro, éste desarrolla agujeros. La textura del cerebro cambia y se vuelve como una esponja de cocina.
Galería de imágenes
6 Imágenes




Formas y causas
- ECJ esporádica: Es la forma más frecuente (aprox. 85–90% de los casos). Aparece sin causa conocida, posiblemente por el plegamiento espontáneo de la proteína priónica.
- ECJ familiar (hereditaria): Causada por mutaciones en el gen PRNP. Suele aparecer en familias con varios miembros afectados y puede presentarse a edades más tempranas.
- ECJ iatrogénica: Transmisión accidental mediante procedimientos médicos, por ejemplo, instrumentos quirúrgicos contaminados, implantes de duramadre o administración de hormonas de crecimiento derivadas de cadáveres infectados (fenómenos raros hoy día).
- ECJ variante (vCJD): Relacionada con la exposición a carne contaminada por EEB (enfermedad de las vacas locas). Esta forma es menos frecuente y afectó a personas más jóvenes durante los brotes asociados a BSE.
Cómo se transmiten los priones
Los priones no contienen material genético como virus o bacterias; son proteínas mal plegadas que inducen a otras proteínas normales a plegarse de forma anómala. Son muy resistentes a métodos habituales de desinfección (calor, radiación, muchos desinfectantes químicos). Por ello, la prevención en ámbitos sanitarios requiere protocolos especiales de esterilización y manejo de materiales de riesgo.
Síntomas y evolución
La ECJ progresa con rapidez en comparación con otras enfermedades neurodegenerativas. Los síntomas típicos incluyen:
- Demencia progresiva: pérdida rápida de la memoria, confusión, alteraciones del pensamiento y del juicio.
- Mioclonías: sacudidas musculares bruscas e involuntarias, frecuentes en ECJ.
- Alteraciones del habla y la visión: dificultad para hablar, pérdida de visión o alucinaciones visuales.
- Ataxia y problemas de coordinación: dificultad para caminar y para realizar movimientos finos.
- Signos neurológicos variados: rigidez, temblor, alteraciones del sueño y cambios conductuales o psiquiátricos.
El curso de la enfermedad suele ser fulminante; la mayoría de las personas fallecen en meses a pocos años desde el inicio de los síntomas. La supervivencia media en la ECJ es de alrededor de 6 a 12 meses tras el diagnóstico en la forma esporádica.
Diagnóstico
El diagnóstico es clínico y apoya en pruebas complementarias:
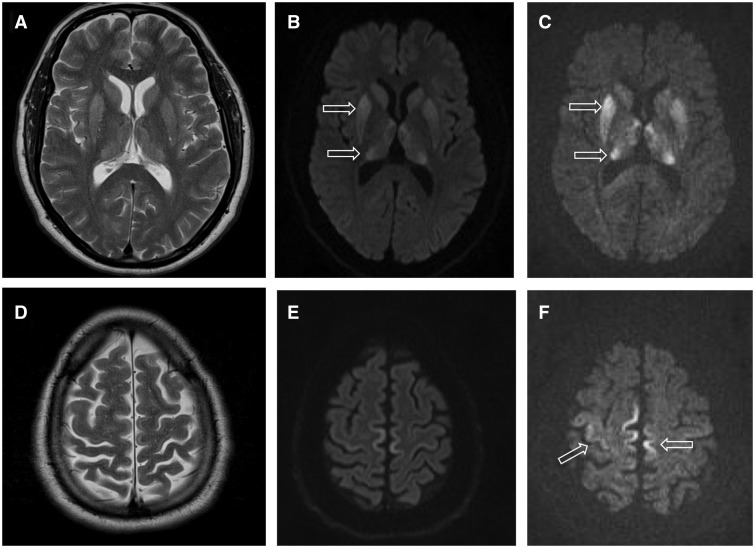
- Resonancia magnética (RM): cambios característicos en córtex y ganglios basales (hiperseñal en DWI/FLAIR).
- Electroencefalograma (EEG): puede mostrar complejos periódicos en algunas etapas de la enfermedad.
- Análisis del líquido cefalorraquídeo (LCR): marcadores como 14-3-3, tau elevados y, en la actualidad, la prueba RT-QuIC (real-time quaking-induced conversion) aumenta la sensibilidad y especificidad para detectar priones.
- Biopsia o autopsia cerebral: la confirmación definitiva suele requerir examen neuropatológico postmortem o, en casos seleccionados, biopsia cerebral.
Tratamiento y manejo
No existe tratamiento curativo ni antiviral eficaz para la ECJ. El manejo se centra en cuidados de apoyo y control de síntomas:
- Control de la mioclonía y de la ansiedad con medicación (por ejemplo, anticonvulsivantes o benzodiacepinas cuando proceda).
- Manejo del dolor, la alimentación y las complicaciones médicas (infecciones, problemas respiratorios).
- Cuidados paliativos y apoyo a la familia y cuidadores, planificación anticipada y medidas de confort.
- En casos hereditarios, asesoramiento genético para familiares en riesgo.
Prevención y control
- Rigurosos protocolos de esterilización y eliminación de material contaminado en centros sanitarios.
- Evitar el uso de tejidos de alto riesgo (tejido cerebral, retina, médula espinal) en procedimientos cuando no se puede garantizar la ausencia de priones.
- En controles veterinarios y de seguridad alimentaria, reducir la exposición humana a animales infectados (medidas aplicadas ante brotes de EEB/BSE).
- Vigilancia epidemiológica y notificación de casos para controlar fuentes iatrogénicas y brotes.
Epidemiología
Es una enfermedad rara: la incidencia global es de aproximadamente 1 a 2 casos por millón de habitantes por año. La forma esporádica afecta sobre todo a adultos mayores (edad media alrededor de 60 años), mientras que la vCJD se observó en personas más jóvenes durante los brotes relacionados con la EEB.
Conclusión
La enfermedad de Creutzfeldt-Jakob es una enfermedad priónica grave, progresiva y letal. Aunque es poco frecuente, sus características (rápida progresión, dificultad diagnóstica y resistencia de los priones a la desinfección) la convierten en un problema clínico y de salud pública que exige diagnóstico rápido, medidas de control estrictas y apoyo integral a los pacientes y sus familias.
Tipos y causas de la ECJ
Los tipos de ECJ incluyen:
- variante (vCJD):
Este tipo de ECJ puede ser causada por la ingesta de alimentos con priones, como la carne de vacas con EEB ("enfermedad de las vacas locas"). Sin embargo, esta es una causa muy poco común de la ECJ.
- esporádica (sCJD):
Es el tipo más común de ECJ. El 85% de los casos de ECJ son esporádicos. Nadie sabe cuál es la causa de la ECJ esporádica; parece ocurrir al azar.
- familiar (fCJD):
La mayor parte del otro 15% de los casos de ECJ son de tipo familiar. Se trata de una forma de ECJ hereditaria.
- iatrogénico:
Esta forma de ECJ suele estar causada por un procedimiento médico en el que una persona recibe sangre o tejidos de alguien con ECJ. Por ejemplo, una persona puede contraer la ECJ iatrogénica si recibe una transfusión de sangre o un trasplante de córnea de alguien que tiene la ECJ.
Signos y síntomas
El primer síntoma de la enfermedad de Creutzfeldt-Jakob es la demencia, que empeora muy rápidamente. La demencia provoca pérdida de memoria, cambios de personalidad y alucinaciones.
Otros síntomas mentales comunes son:
- Ansiedad
- Depresión
- Paranoia
- Síntomas obsesivo-compulsivos
- Psicosis
Los síntomas físicos de la ECJ suelen incluir:
- Problemas para hablar
- Movimientos espasmódicos (mioclonía)
- Problemas de equilibrio (ataxia)
- Problemas para caminar
- Temblores o rigidez
- Problemas de visión
- Problemas para tragar, lo que puede dificultar o imposibilitar la alimentación
- Problemas para toser, que pueden causar neumonía
- Movimientos que el paciente no puede controlar (discinesia)
La mayoría de los enfermos de ECJ mueren en los seis meses siguientes a la aparición de los primeros síntomas. A menudo, mueren de neumonía causada por problemas de tos. Alrededor del 15% de los pacientes sobreviven dos o más años. Algunos pacientes han vivido entre 4 y 5 años con síntomas principalmente mentales hasta que la enfermedad empeora y provoca más síntomas físicos. Una vez que esto ocurre, la gente suele morir en el plazo de un año.
Los síntomas de la enfermedad de Creutzfeldt-Jakob se deben a la muerte de un número cada vez mayor de células nerviosas del cerebro. Cuando los científicos observan el tejido cerebral de un paciente con ECJ al microscopio, pueden ver muchos agujeros diminutos en los que han muerto zonas enteras de células nerviosas.
Diagnóstico
Los médicos pueden sospechar la existencia de la ECJ cuando una persona presenta determinados síntomas. Por ejemplo, la demencia suele empeorar lentamente. La demencia que empeora muy rápidamente es inusual. Junto con síntomas como los movimientos espasmódicos, estos síntomas pueden apuntar a una posible ECJ.
A continuación, se pueden realizar pruebas para demostrar si la persona tiene la ECJ. Estas pruebas incluyen:
- Electroencefalografía (EEG): Esta prueba muestra la actividad eléctrica del cerebro. A menudo, el médico podrá ver cambios en el EEG que son comunes en las personas con ECJ. El tipo de cambios que aparecen en el EEG dependerá del tipo de ECJ que tenga el paciente y de lo avanzada que esté su enfermedad.
- Punción lumbar (punción espinal): Esta prueba permite estudiar el líquido cefalorraquídeo (el líquido que rodea el cerebro y la médula espinal), buscando una proteína específica ("proteína 14-3-3")
- Resonancia magnética del cerebro: Prueba que utiliza un imán muy potente para tomar imágenes del cerebro.
- Biopsia: Para realizar una biopsia, un cirujano utiliza una aguja para extraer un pequeño trozo de tejido del cuerpo, de modo que los médicos puedan examinarlo al microscopio. La vECJ puede diagnosticarse con una biopsia de las amígdalas. Para todos los demás tipos de ECJ, una biopsia del cerebro es la única manera de saber con seguridad si una persona tiene ECJ. Sin embargo, dado que una biopsia del cerebro puede causar daños cerebrales, normalmente no se realiza una biopsia cerebral si otras pruebas ya han demostrado que una persona probablemente tiene la ECJ.

Tratamiento
A fecha de 2016, no existe ningún tratamiento que cure la ECJ o incluso que ralentice sus efectos. Se están realizando muchos experimentos para tratar de encontrar tratamientos.
En la actualidad, los únicos tratamientos para la ECJ son medicamentos que tratan los síntomas de la enfermedad y ayudan a los pacientes a estar más cómodos. Por ejemplo, los pacientes que sufren convulsiones pueden recibir medicamentos anticonvulsivos. Las benzodiacepinas pueden hacer que las sacudidas musculares sean menos frecuentes.
Los pacientes también pueden optar por someterse a procedimientos médicos que les ayuden a paliar los malos síntomas. Por ejemplo, la ECJ puede causar tantos problemas para tragar que la persona no puede comer. Algunas personas con ECJ optan por colocarse una sonda de alimentación cuando ya no pueden comer. Se trata de una sonda que se introduce en el estómago, de modo que se puede administrar un líquido especial directamente en el estómago para nutrir a la persona.
Páginas relacionadas
- Prion
- Enfermedad de los priones
- Enfermedad terminal
Preguntas y respuestas
P: ¿Qué es la enfermedad de Creutzfeldt-Jakob?
R: La enfermedad de Creutzfeldt-Jakob (ECJ) es una enfermedad neurológica degenerativa, incurable y siempre mortal.
P: ¿Existe cura para la ECJ?
R: No, no existe cura para la ECJ.
P: ¿Por qué a veces se hace referencia a la ECJ como una forma humana de la "enfermedad de las vacas locas"?
R: La ECJ se denomina a veces una forma humana de la "enfermedad de las vacas locas" porque la encefalopatía espongiforme bovina (EEB), que es la causa de un tipo raro de ECJ, se conoce comúnmente como "enfermedad de las vacas locas".
P: ¿Cuál es la causa de la ECJ?
R: La ECJ está causada por un agente infeccioso llamado prión, que es una proteína que se pliega incorrectamente y puede hacer copias de sí misma cambiando las proteínas correctamente plegadas por otras mal plegadas.
P: ¿Qué le ocurre al tejido cerebral en la ECJ?
R: La enfermedad de Creutzfeldt-Jakob hace que el tejido cerebral se vuelva insano muy rápidamente, lo que provoca la aparición de agujeros en el cerebro y un cambio en la textura del cerebro que se vuelve como una esponja de cocina.
P: ¿La EEB es la misma enfermedad que la ECJ?
R: No, la EEB no es la misma enfermedad que la ECJ; en realidad es la causa de un tipo raro de ECJ.
P: ¿Cómo causan la ECJ los priones?
R: Los priones causan la ECJ al plegarse incorrectamente y hacer copias de sí mismos a expensas de las proteínas correctamente plegadas en el cerebro. Esto provoca la destrucción del tejido cerebral sano y el desarrollo de los agujeros característicos de la enfermedad.
Artículos relacionados
Autor
AlegsaOnline.com Enfermedad de Creutzfeldt-Jakob: definición, causas (priones) y síntomas Leandro Alegsa
URL: https://es.alegsaonline.com/art/24152
Fuentes
- cdc.gov : "CJD (Creutzfeldt–Jakob Disease, Classic)"
- ncbi.nlm.nih.gov : "Creutzfeldt–Jakob disease: Transmissible spongiform encephalopathy; vCJD; CJD; Jacob-Creutzfeldt disease"
- bmj.com : "Bovine spongiform encephalopathy and variant Creutzfeldt–Jakob disease"
- doi.org : 10.1101/SQB.1996.061.01.052
- pubmed.ncbi.nlm.nih.gov : 9246478
- www3.interscience.wiley.com : "MM2-cortical-type sporadic Creutzfeldt–Jakob disease with early stage cerebral cortical pathology presenting with a rapidly progressive clinical course"
- doi.org : 10.1111/j.1440-1789.2008.00904.x
- pubmed.ncbi.nlm.nih.gov : 18410280
- who.int : who.int: "Fact sheets no 180: Variant Creutzfeldt-Jakob disease" Feb 2012 ed.
- prion.ucl.ac.uk : "Sporadic Prion Disease"
- doi.org : 10.1056/NEJM198610163151605
- pubmed.ncbi.nlm.nih.gov : 3762620
- prion.ucl.ac.uk : "Sporadic Prion Disease"
- merckmanuals.com : "Creutzfeldt–Jakob Disease (CJD)"
- ncbi.nlm.nih.gov : "Creutzfeldt-Jakob disease: Updated diagnostic criteria, treatment algorithm, and the utility of brain biopsy"