Hipertensión pulmonar: causas, síntomas, diagnóstico y tratamiento
Hipertensión pulmonar: conoce causas, síntomas, diagnóstico y tratamientos efectivos. Aprende a detectar y manejar la enfermedad para mejorar la respiración y la calidad de vida.
La hipertensión pulmonar o HP es una afección en la que hay una presión arterial elevada en los pulmones. Esta afección dificulta la respiración. Algunas personas que la padecen necesitan más oxígeno. Esta afección también puede hacer que una persona se maree y se canse fácilmente. Algunas personas con esta enfermedad se desmayan fácilmente. Los síntomas empeoran al hacer ejercicio o trabajar duro. La hipertensión pulmonar es una enfermedad grave que puede ser mortal. Esta enfermedad dificulta el bombeo de sangre por parte del corazón. Como el corazón tiene que trabajar más, también puede enfermar. Algunas personas muy enfermas pueden necesitar un trasplante de pulmón o un trasplante cardiopulmonar para vivir. El nombre completo de la hipertensión pulmonar es hipertensión arterial pulmonar, aunque la mayoría de la gente la llama pah, ph o pha.
Galería de imágenes
9 Imágenes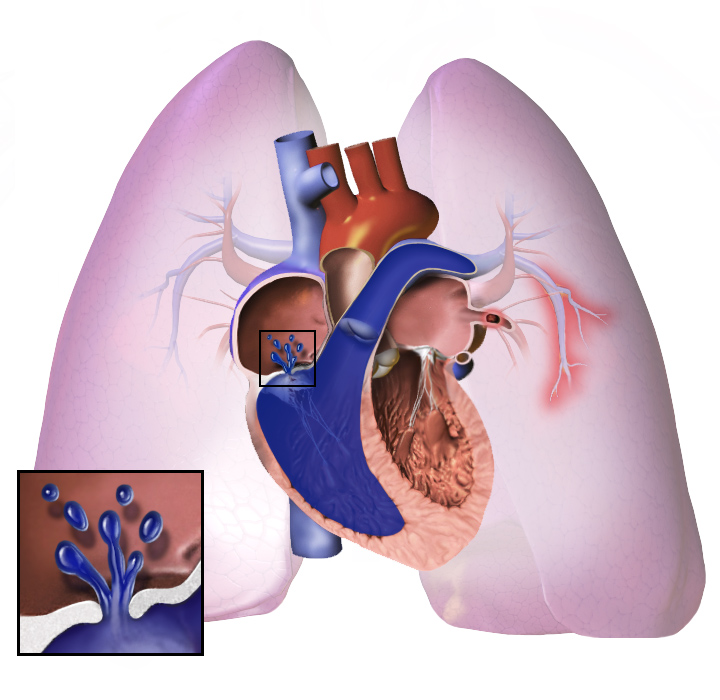






Causas y mecanismos
- Hipertensión arterial pulmonar idiopática o hereditaria: sin causa clara o por mutaciones genéticas (por ejemplo, BMPR2).
- Asociada a otras enfermedades: enfermedades del tejido conectivo (esclerodermia), infección por VIH, enfermedad hepática (hipertensión portal), uso de drogas o tóxicos (anfetaminas, anorexígenos), cardiopatías congénitas con cortocircuito izquierda-derecha.
- Enfermedades pulmonares y/o hipoxia crónica: EPOC, enfermedad intersticial, apnea del sueño.
- Tromboembólica crónica: obstrucción crónica de las arterias pulmonares por émbolos organizados (CTEPH).
Los mecanismos que aumentan la presión incluyen vasoconstricción persistente, remodelado vascular (engrosamiento y fibrosis de las paredes arteriales), trombosis in situ e inflamación.
Síntomas
- Disnea de esfuerzo progresiva (lo más frecuente).
- Fatiga y disminución de la capacidad para realizar actividades cotidianas.
- Dolor torácico o sensación de opresión.
- Mareos o síncope, sobre todo con el esfuerzo.
- Palpitaciones o arritmias.
- Signos de insuficiencia cardiaca derecha: hinchazón de piernas (edema), acumulación de líquido abdominal, pérdida de apetito.
- Coloración azulada de labios o uñas (cianosis) en casos avanzados.
Diagnóstico
El diagnóstico se basa en la sospecha clínica y en pruebas que evalúan la presión y función del sistema pulmonar y del corazón derecho:
- Exploración física: ruidos cardiacos alterados (fuerte segundo ruido pulmonar), soplos, signos de congestión sistémica.
- Pruebas no invasivas: radiografía de tórax, electrocardiograma (ECG), ecocardiograma transtorácico (estimación de presiones y función del ventrículo derecho), pruebas de función pulmonar y oximetría o gasometría arterial.
- Biomarcadores: péptidos natriuréticos (BNP/NT-proBNP) para valorar sobrecarga del corazón derecho.
- Pruebas de imagen avanzadas: tomografía computarizada (TAC) y gammagrafía ventilación-perfusión (V/Q) —fundamental para detectar hipertensión tromboembólica crónica (CTEPH).
- Cateterismo cardiaco derecho: es la prueba de referencia (gold standard) para confirmar la hipertensión pulmonar, medir presiones y resistencia vascular pulmonar, y realizar test de vasorreactividad en caso necesario.
- Otras pruebas: pruebas de esfuerzo (6 minutos), estudios de sueño, serologías y análisis para enfermedades asociadas (inmunología, VIH, función hepática).
Tratamiento
El manejo depende de la causa, la gravedad y la respuesta al tratamiento. Requiere a menudo un equipo multidisciplinario (cardiólogo, neumólogo, reumatólogo, unidades de hipertensión pulmonar).
- Medidas generales: oxigenoterapia en hipoxemia, diuréticos para controlar edemas, restricción de sal cuando esté indicada, vacunación (gripe, neumococo), rehabilitación cardiopulmonar y consejo sobre actividad física.
- Tratamiento específico farmacológico:
- Antagonistas del receptor de endotelina (bosentan, ambrisentan).
- Inhibidores de la fosfodiesterasa tipo 5 (sildenafil, tadalafil).
- Análogos de prostaciclina y prostanoides (epoprostenol, treprostinil, iloprost) —especialmente para formas avanzadas.
- Estimuladores de la guanilato ciclasa soluble (riociguat) —útil en CTEPH e HAP en ciertos casos.
- Bloqueadores de canales de calcio orales —solo en pacientes que muestran vasorreactividad positiva en el cateterismo.
- Anticoagulación: puede indicarse en determinados casos (por ejemplo, CTEPH o cuando hay riesgo tromboembólico).
- Tratamientos invasivos y quirúrgicos:
- Endarterectomía pulmonar (pulmonary thromboendarterectomy) para CTEPH operable —potencialmente curativa.
- Angioplastia o balón pulmonar (balloon pulmonary angioplasty) para CTEPH inoperable.
- Septostomía auricular como puente paliativo en casos seleccionados con hipertensión pulmonar grave.
- Trasplante: trasplante de pulmón o trasplante cardiopulmonar en pacientes refractarios a tratamiento médico.
Seguimiento y pronóstico
El pronóstico varía según la causa y la rapidez del diagnóstico y tratamiento. Gracias a los tratamientos actuales la supervivencia y la calidad de vida han mejorado, pero la hipertensión pulmonar puede ser progresiva. El seguimiento incluye valoración clínica periódica, pruebas funcionales (6 minutos), ecocardiogramas y ajuste terapéutico según respuesta.
Prevención y consejos prácticos
- Controlar y tratar enfermedades de base (enfermedad pulmonar, cardiopatías, enfermedades autoinmunes).
- Evitar fármacos y sustancias asociadas a riesgo (consultar con el médico).
- Dejar de fumar y mantener un peso adecuado.
- Vacunarse y evitar infecciones respiratorias que puedan empeorar la función pulmonar.
- Buscar atención médica si aparecen dificultad respiratoria progresiva, síncope u otros síntomas nuevos.
Cuándo acudir de urgencia
- Síncope o desmayo repentino.
- Aumento súbito de la dificultad para respirar o dolor torácico intenso.
- Edema rápido o dolor abdominal intenso por congestión hepática.
Si sospecha hipertensión pulmonar, es importante consultar con su médico para evaluación y derivación a una unidad especializada. Un diagnóstico y tratamiento tempranos mejoran las opciones terapéuticas y la calidad de vida.
Signos y síntomas
Las personas con hipertensión pulmonar tienen dificultades para respirar. También se cansan con facilidad. Algunas de ellas también se desmayan con facilidad. Pueden tener dolor en el pecho. Algunos pacientes presentan hinchazón de pies y tobillos. Estos síntomas empeoran durante el ejercicio o el trabajo duro.
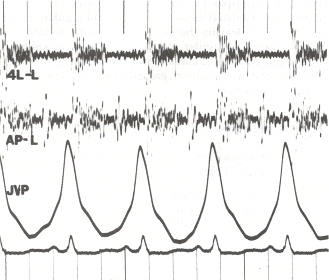
Como muchas enfermedades pueden dificultar la respiración, el médico debe conocer los antecedentes del paciente. Esto ayuda al médico a tratar al paciente, incluso si éste tiene otra enfermedad. El médico también realiza varias pruebas. La hipertensión pulmonar hace que el corazón suene diferente. Una de las pruebas consiste en medir la presión sanguínea dentro de la arteria pulmonar, el vaso sanguíneo que va del corazón a los pulmones.
Para establecer la causa, el médico suele realizar una historia clínica completa. Se toman los antecedentes familiares detallados para determinar si la enfermedad puede ser familiar. Se consideran significativos los antecedentes de exposición a la cocaína, a la metanfetamina, al alcohol, que conducen a la cirrosis, y al tabaquismo, que conducen al enfisema. La exploración física se realiza en busca de los signos típicos de la hipertensión pulmonar, como un P2 fuerte (sonido de cierre de la válvula pulmonar), un oleaje (para)esternal, distensión venosa yugular, edema pediátrico, ascitis, reflujo hepatoyugular, palos, etc.
Lo que va mal en el cuerpo
En la hipertensión pulmonar, los vasos sanguíneos de los pulmones se estrechan demasiado. La presión sanguínea en los pulmones se eleva. El corazón trabaja mucho para bombear la sangre a través de los vasos sanguíneos estrechos. Más adelante, los vasos sanguíneos de los pulmones se vuelven duros y gruesos. El corazón debe trabajar más.
El corazón puede trabajar tanto que se enferma. Esto se denomina insuficiencia cardíaca. El corazón enfermo no puede bombear bien la sangre. Llega menos sangre a los pulmones, por lo que la sangre recibe menos oxígeno. Esto dificulta la respiración. Esto empeora cuando se hace ejercicio o se trabaja mucho.
Causa
La causa más común de la hipertensión pulmonar es la insuficiencia cardíaca izquierda. Esto provoca una hipertensión venosa pulmonar. Esto conduce a un edema pulmonar, o acumulación de líquido en los pulmones.
Muchas enfermedades pueden causar hipertensión arterial pulmonar (HAP).
- Enfermedades pulmonares que hacen que la sangre tenga menos oxígeno, como:
· enfermedad pulmonar obstructiva crónica o EPOC
· enfermedad pulmonar intersticial
· Síndrome de Pickwick
- problemas del sistema inmunológico, como:
· SIDA
· esclerodermia
· otros trastornos autoinmunes
- problemas de hígado
· cirrosis
· hipertensión portal
- otras causas
· apnea del sueño
· tomar pastillas para perder peso, como Fen-Phen, Aminorex, fenfluramina (Pondimin) y fentermina
· enfermedad de células falciformes,
· enfermedad cardíaca congénita
· enfermedades de la tiroides,
· tomar drogas como la cocaína
· posiblemente Herpesvirus humano 8
Cuando una persona tiene hipertensión pulmonar sin ninguna otra causa, se denomina hipertensión arterial pulmonar idiopática o HAPI.
Cuando existen antecedentes familiares, la enfermedad se denomina hipertensión arterial pulmonar familiar (HAPF). Actualmente se considera que la HIP y la HAPF son trastornos genéticos relacionados con mutaciones en el gen BMPR2, que codifica un receptor para las proteínas morfogenéticas óseas, así como en el gen 5-HT(2B), que codifica un receptor de serotonina.
En medicina, la hipertensión pulmonar (HP) es un aumento de la presión sanguínea en la arteria pulmonar o en la vasculatura pulmonar, que provoca falta de aire, mareos, desmayos y otros síntomas, todos ellos exacerbados por el esfuerzo. Dependiendo de la causa, la hipertensión pulmonar puede ser una enfermedad grave con una marcada disminución de la tolerancia al ejercicio y una insuficiencia cardíaca derecha. Fue identificada por primera vez por el Dr. Ernst von Romberg en 1891. Puede ser de cinco tipos diferentes: arterial, venosa, hipóxica, tromboembólica o miscelánea.
Aunque los términos hipertensión pulmonar primaria (que significa de causa desconocida) e hipertensión pulmonar secundaria (que significa debida a otra condición médica) todavía persisten en los materiales difundidos a los pacientes y al público en general, estos términos se han abandonado en gran medida en la literatura médica. Este cambio se ha producido porque la antigua clasificación dicotómica no reflejaba la fisiopatología ni el resultado. Llevaba a decisiones terapéuticas erróneas, es decir, a tratar únicamente la hipertensión pulmonar "primaria". Esto, a su vez, llevó al nihilismo terapéutico a muchos pacientes etiquetados como hipertensión pulmonar "secundaria", y podría haber contribuido a su muerte. El término "hipertensión pulmonar primaria" se ha sustituido ahora por el de "hipertensión arterial pulmonar idiopática". Los términos "hipertensión pulmonar primaria" y "secundaria" ya no deben utilizarse. Encontrará más detalles en la sección de Clasificación.
Causa
La causa más común de la hipertensión pulmonar es la insuficienciacardíaca izquierda que da lugar a una hipertensión venosa pulmonar. Puede deberse a un mal funcionamiento sistólico o diastólico del ventrículo izquierdo o a una disfunción valvular, como la regurgitación mitral o la estenosis mitral. Suele manifestarse como un edema pulmonar.
Las causas más comunes de la hipertensión arterial pulmonar (HAP) son el VIH, la esclerodermia y otros trastornos autoinmunes, la cirrosis y la hipertensión portal, la anemia de células falciformes, las cardiopatías congénitas y las enfermedades tiroideas, entre otras. El uso de píldoras para adelgazar, como Fen-Phen, Aminorex, fenfluramina (Pondimin) y fentermina, condujo al desarrollo de HAP en el pasado.
Patogénesis
Cualquiera que sea la causa inicial, la hipertensión pulmonar implica el estrechamiento de los vasos sanguíneos conectados a los pulmones y dentro de ellos. Esto dificulta que el corazón bombee la sangre a través de los pulmones, del mismo modo que es más difícil hacer que el agua fluya a través de una tubería estrecha que de una ancha. Con el tiempo, los vasos sanguíneos afectados se vuelven más rígidos y gruesos, aumentando aún más la presión sanguínea dentro de los pulmones y dificultando el flujo sanguíneo. Además, el aumento de la carga de trabajo del corazón provoca el engrosamiento y el agrandamiento del ventrículo derecho, lo que hace que el corazón sea menos capaz de bombear sangre a través de los pulmones, provocando una insuficiencia cardíaca derecha. Al disminuir el flujo de sangre a través de los pulmones, el lado izquierdo del corazón recibe menos sangre. Esta sangre también puede transportar menos oxígeno de lo normal. Por lo tanto, al lado izquierdo del corazón le resulta cada vez más difícil bombear para suministrar suficiente oxígeno al resto del cuerpo, especialmente durante la actividad física.
Diagnóstico
Dado que la hipertensión pulmonar puede ser de 5 tipos principales, hay que realizar una serie de pruebas para distinguir la hipertensión arterial pulmonar de las variedades venosa, hipóxica, tromboembólica o miscelánea.
Se realiza una exploración física para buscar los signos típicos de la hipertensión pulmonar. Entre ellos se encuentran las alteraciones de los ruidos cardíacos, como una S muy dividida 2o segundo ruido cardíaco, una P fuerte2 o sonido de cierre de la válvula pulmonar (parte del segundo ruido cardíaco), un oleaje (para)esternal, un posible S3 o tercer ruido cardíaco y regurgitación pulmonar. Otros signos son la distensión venosa yugular (agrandamiento de las venas yugulares), el edema periférico (hinchazón de los tobillos y los pies), la ascitis (hinchazón abdominal debida a la acumulación de líquido), el reflujo hepatoyugular y los golpes.
Se requieren procedimientos adicionales para confirmar la presencia de hipertensión pulmonar y excluir otros posibles diagnósticos. Por lo general, éstos incluyen pruebas de función pulmonar, análisis de sangre, electrocardiograma (ECG), mediciones de gases en sangre arterial, radiografías de tórax (seguidas de una tomografía computarizada de alta resolución si se sospecha de una enfermedad pulmonar intersticial) y exploración de ventilación-perfusión o V/Q para excluir la hipertensión pulmonar tromboembólica crónica. La biopsia de pulmón no suele estar indicada a menos que se piense que la hipertensión pulmonar se debe a una enfermedad pulmonar intersticial subyacente. Sin embargo, las biopsias de pulmón conllevan riesgos de hemorragia debido a la elevada presión sanguínea intrapulmonar. La mejoría clínica suele medirse mediante la "prueba de la marcha de seis minutos", es decir, la distancia que el paciente puede caminar en seis minutos. La estabilidad y la mejora de esta medida se correlacionan con una mayor supervivencia.
Aunque la presión arterial pulmonar puede estimarse sobre la base de la ecocardiografía, el muestreo de presión con un catéter de Swan-Ganz proporciona la medición más definitiva. La PAOP y la RVP no pueden medirse directamente con la ecocardiografía. Por lo tanto, el diagnóstico de la HAP requiere un cateterismo cardíaco. Un catéter de Swan-Ganz también puede medir el gasto cardíaco, que es mucho más importante para medir la gravedad de la enfermedad que la presión arterial pulmonar.
La presión arterial pulmonar normal en una persona que vive a nivel del mar tiene un valor medio de 12-16 mm Hg (1600-2100 Pa). Existe una hipertensión pulmonar definitiva cuando las presiones medias en reposo superan los 25 mm Hg (3.300 Pa). Si la presión arterial pulmonar media se eleva por encima de 30 mm Hg (4000 Pa) con el ejercicio, también se considera hipertensión pulmonar.
El diagnóstico de HAP requiere la presencia de hipertensión pulmonar con otras dos condiciones. La presión de oclusión de la arteria pulmonar (PAOP o PCWP) debe ser inferior a 15 mm Hg (2000 Pa) y la resistencia vascular pulmonar (PVR) debe ser superior a 3 unidades Wood (240 dyn-s-cm −5o 2,4 mN-s-cm −5).
Clasificación
Clasificación actual
En 2003, se convocó en Venecia el Tercer Simposio Mundial sobre Hipertensión Arterial Pulmonar para modificar la clasificación en función de los nuevos conocimientos sobre los mecanismos de la enfermedad. El sistema revisado desarrollado por este grupo proporciona el marco actual para entender la hipertensión pulmonar.
El sistema incluye varias mejoras con respecto al anterior sistema de clasificación de Evian de 1998. Se han actualizado las descripciones de los factores de riesgo y se ha revisado la clasificación de las derivaciones sistémico-pulmonares congénitas. Se recomendó una nueva clasificación de los factores genéticos en la HP, pero no se aplicó porque se consideró que los datos disponibles eran inadecuados.
El sistema de clasificación revisado de Venecia 2003 puede resumirse como sigue:
- Grupo I de la OMS - Hipertensión arterial pulmonar (HAP)
- Grupo II de la OMS - Hipertensión pulmonar asociada a cardiopatía izquierda
- Grupo III de la OMS - Hipertensión pulmonar asociada a enfermedades pulmonares y/o hipoxemia
- Grupo IV de la OMS - Hipertensión pulmonar por enfermedad trombótica y/o embólica crónica
- Grupo V de la OMS - Varios
Terminología anterior
Antiguamente se utilizaban los términos hipertensión pulmonar primaria y secundaria (HPP y HPS) para clasificar la enfermedad. Esto llevó a suponer que sólo debía tratarse la enfermedad primaria, y que la variedad secundaria debía ignorarse en favor de tratar sólo la enfermedad subyacente. De hecho, todas las formas de hipertensión arterial pulmonar son tratables. Desgraciadamente, este sistema de clasificación aún persiste en la mente de muchos médicos, y probablemente hace que a muchos pacientes se les niegue el tratamiento. Este enfoque nihilista de la hipertensión arterial pulmonar también puede contribuir al infradiagnóstico. Se calcula que hay unos 100.000 pacientes con HAP en los Estados Unidos, pero sólo se han diagnosticado entre 15 y 20.000. Muchos otros han sido diagnosticados erróneamente como EPOC, asma o insuficiencia cardíaca congestiva.
El término hipertensión pulmonar primaria (HPP) ha sido sustituido por el de hipertensión arterial pulmonar idiopática (HAPI) en gran parte de la literatura médica. Sin embargo, algunos médicos siguen utilizando la antigua clasificación de forma inapropiada.
Epidemiología
La HIPP es una enfermedad rara con una incidencia de unos 2-3 por millón al año y una prevalencia de unos 15 por millón. Las mujeres tienen casi tres veces más probabilidades de presentar HAPI que los hombres.
Otras formas de HAP son mucho más frecuentes. En la esclerodermia la incidencia se ha estimado entre el 6 y el 60% de todos los pacientes, en la artritis reumatoide hasta el 21%, en el lupus eritematoso sistémico entre el 4 y el 14%, en la hipertensión portal entre el 2 y el 5%, en el VIH alrededor del 0,5% y en la anemia de células falciformes entre el 20 y el 40%.
Las píldoras dietéticas como Fen-Phen produjeron una incidencia anual de 25-50 por millón al año.
Tratamiento
El tratamiento viene determinado por si la HP es arterial, venosa, hipóxica, tromboembólica o miscelánea. Dado que la hipertensión venosa pulmonar es sinónimo de insuficiencia cardíaca congestiva, el tratamiento consiste en optimizar la función ventricular izquierda mediante el uso de diuréticos, betabloqueantes, inhibidores de la ECA, etc., o en reparar/sustituir la válvula mitral o la válvula aórtica.
En la HAP, los cambios en el estilo de vida, la digoxina, los diuréticos, los anticoagulantes orales y la oxigenoterapia se consideran un tratamiento convencional, pero nunca se ha demostrado que sean beneficiosos de forma aleatoria y prospectiva.
Los antagonistas del calcio en dosis elevadas son útiles sólo en el 5% de los pacientes con HAPI que son vasorreactivos por catéter de Swan-Ganz. Lamentablemente, los antagonistas del calcio se han utilizado en gran medida de forma incorrecta, prescribiéndose a muchos pacientes con HAP no vasorreactiva, lo que ha provocado un exceso de morbilidad y mortalidad.
Sustancias vasoactivas
Hay tres vías principales que intervienen en la proliferación y contracción anormales de las células del músculo liso de la arteria pulmonar en los pacientes con hipertensión arterial pulmonar. Estas vías corresponden a importantes objetivos terapéuticos en esta enfermedad y desempeñan un papel en la determinación de cuál de las tres clases de fármacos -antagonistas de los receptores de endotelina, inhibidores de la fosfodiesterasa tipo 5 y derivados de la prostaciclina- se utilizará.
La prostaciclina (prostaglandina I2 ) se considera habitualmente el tratamiento más eficaz para la HAP. El epoprostenol (prostaciclina sintética, comercializada como Flolan®) se administra mediante una infusión continua que requiere un catéter venoso central semipermanente. Este sistema de administración puede provocar sepsis y trombosis. Flolan® es inestable, por lo que debe mantenerse en hielo durante su administración. Como tiene una vida media de 3 a 5 minutos, la infusión debe ser continua (24/7), y su interrupción puede ser fatal. Por ello, se han desarrollado otros prostanoides. El treprostinil (Remodulin®) puede administrarse por vía intravenosa o subcutánea, pero la forma subcutánea puede ser muy dolorosa. El iloprost (Ilomedin®) también se utiliza en Europa por vía intravenosa y tiene una vida media más larga. El iloprost (comercializado como Ventavis®) es la única forma inhalada de prostaciclina aprobada para su uso en Estados Unidos y Europa. Esta forma de administración tiene la ventaja de que se deposita selectivamente en los pulmones con menos efectos secundarios sistémicos.
El antagonista del receptor de la endotelina dual (ET Ay ETB ) bosentán (comercializado como Tracleer®) fue aprobado en 2001. Dos antagonistas selectivos de los receptores de endotelina (Asólo ET) están en las últimas fases de aprobación: sitaxsentan y ambrisentan. El sildenafilo, un inhibidor selectivo de la fosfodiesterasa tipo 5 (PDE5) específica del GMPc, se aprobó para el tratamiento de la HAP en 2005. Se comercializa para la HAP como Revatio®. El tadalafilo (comercializado actualmente como Cialis® para la disfunción eréctil) se encuentra actualmente en fase III de ensayos. El péptido intestinal vasoactivo por inhalación debería entrar en ensayos clínicos para la HAP en 2007. El PRX-08066 es un antagonista de la serotonina que se está desarrollando para la hipertensión pulmonar hipóxica.
Quirúrgico
La septostomía auricular es un procedimiento quirúrgico que crea una comunicación entre las aurículas derecha e izquierda. Alivia la presión en el lado derecho del corazón, pero a costa de reducir los niveles de oxígeno en la sangre (hipoxia). Se realiza mejor en centros con experiencia. El trasplante de pulmón cura la hipertensión arterial pulmonar, pero deja al paciente con las complicaciones del trasplante y una supervivencia de unos 5 años.
La tromboendarterectomía pulmonar (TEP) es un procedimiento quirúrgico que se utiliza para la hipertensión pulmonar tromboembólica crónica. Consiste en la extirpación quirúrgica de un trombo organizado (coágulo) junto con el revestimiento de la arteria pulmonar; es un procedimiento amplio y muy difícil que actualmente se realiza en unos pocos centros seleccionados. Las series de casos muestran un éxito notable en la mayoría de los pacientes.
No se ha establecido un tratamiento para las variedades hipóxicas y misceláneas de la hipertensión pulmonar. Sin embargo, en la actualidad hay estudios de varios agentes que están reclutando pacientes. Muchos médicos tratarán estas enfermedades con los mismos medicamentos que para la HAP, hasta que se disponga de mejores opciones.
Pronóstico
El registro de IPAH de los NIH de la década de 1980 mostraba una mediana de supervivencia sin tratamiento de 2 a 3 años desde el momento del diagnóstico, siendo la causa de muerte generalmente la insuficiencia ventricular derecha (cor pulmonale). Aunque esta cifra es muy citada, probablemente sea irrelevante hoy en día. Los resultados han cambiado drásticamente en las últimas dos décadas. Esto puede deberse a un nuevo tratamiento farmacológico, a una mejor atención general y a un diagnóstico más temprano (sesgo de tiempo de espera). Un estudio reciente sobre los resultados de los pacientes que habían iniciado el tratamiento con bosentan (Tracleer®) mostró que el 86% de los pacientes estaban vivos a los 3 años. Ahora que se dispone de múltiples agentes, se utiliza cada vez más la terapia combinada. No se conoce el impacto de estos agentes en la supervivencia, ya que muchos de ellos se han desarrollado recientemente. No sería descabellado esperar que la mediana de supervivencia supere los 10 años en un futuro próximo.
Preguntas y respuestas
P: ¿Qué es la hipertensión pulmonar o HP?
R: La hipertensión pulmonar o HP es una enfermedad en la que existe una presión arterial elevada en los pulmones.
P: ¿Cuáles son los síntomas de la hipertensión pulmonar?
R: Los síntomas de la hipertensión pulmonar incluyen dificultad para respirar, mareos, fatiga y desmayos.
P: ¿Por qué algunas personas con hipertensión pulmonar necesitan más oxígeno?
R: Algunas personas con hipertensión pulmonar necesitan oxígeno extra porque la enfermedad les dificulta la respiración.
P: ¿Cuándo empeoran los síntomas de la hipertensión pulmonar?
R: Los síntomas de la hipertensión pulmonar empeoran al hacer ejercicio o trabajar mucho.
P: ¿Por qué la hipertensión pulmonar es una enfermedad grave?
R: La hipertensión pulmonar es una afección grave porque dificulta el bombeo de sangre por el corazón y puede ser mortal.
P: ¿Cuál es el nombre completo de la hipertensión pulmonar?
R: El nombre completo de la hipertensión pulmonar es hipertensión arterial pulmonar aunque la mayoría de la gente la llama pah, ph o pha.
P: ¿Qué pueden necesitar algunas personas muy enfermas con hipertensión pulmonar para vivir?
R: Algunas personas muy enfermas con hipertensión pulmonar pueden necesitar un trasplante de pulmón o un trasplante cardiopulmonar para vivir.
Artículos relacionados
Autor
AlegsaOnline.com Hipertensión pulmonar: causas, síntomas, diagnóstico y tratamiento Leandro Alegsa
URL: https://es.alegsaonline.com/art/80022
Fuentes
- ncbi.nlm.nih.gov : PMID 8692238
- ncbi.nlm.nih.gov : PMID 14985486
- ncbi.nlm.nih.gov : PMID 10555089
- ncbi.nlm.nih.gov : PMID 13679525
- ncbi.nlm.nih.gov : PMID 10903931
- ncbi.nlm.nih.gov : PMID 14659797
- ncbi.nlm.nih.gov : PMID 15194171