Osteogénesis imperfecta (huesos frágiles): definición, tipos y síntomas
Osteogénesis imperfecta: guía completa sobre causas genéticas, tipos, síntomas y manejo de huesos frágiles. Conoce diagnóstico, riesgos y prevención de fracturas.
La osteogénesis imperfecta (OI), conocida también como enfermedad de los huesos frágiles, es un trastorno del tejido conectivo de origen genético que provoca fragilidad ósea y tendencia a fracturas con traumatismos leves o incluso espontáneas. Afecta principalmente la producción y la estructura del colágeno tipo I, una proteína esencial que da resistencia al hueso y a otros tejidos. La OI puede presentarse en grados muy variables, desde formas leves con pocas fracturas hasta formas graves que afectan la supervivencia y la calidad de vida.
Galería de imágenes
10 Imágenes







Causas y patrón de herencia
La causa más frecuente son mutaciones en los genes que codifican las cadenas del colágeno tipo I (por ejemplo, COL1A1 y COL1A2), que alteran la cantidad o la calidad del colágeno producido. Aunque con frecuencia la enfermedad se transmite con un patrón autosómico dominante (es decir, basta con que uno de los padres aporte el gen alterado para que el hijo esté afectado), también existen formas autosómicas recesivas y variantes de aparición nueva (mutaciones de novo) en personas sin antecedentes familiares. Actualmente se han identificado muchos genes asociados a fenotipos similares a la OI, lo que explica su gran variabilidad clínica.
Tipos y características clínicas
La clasificación clásica (Sillence) divide la enfermedad en cuatro tipos principales, aunque hoy en día se reconocen más subtipos y variedades genéticas:
- Tipo I (leve): es el más frecuente. Fracturas óseas con traumatismos leves, estatura normal o ligeramente reducida, escleróticas azules o pálidas, y con frecuencia dientes afectados (dentinogénesis imperfecta). Puede aparecer pérdida de audición a partir de la adolescencia o edad adulta.
- Tipo II (perinatalmente letal): forma más grave; muchos fetos presentan fracturas intrauterinas, deformidades óseas marcadas y, con frecuencia, fallecen en el período perinatal o en la primera infancia por complicaciones respiratorias.
- Tipo III (severo y progresivo): fracturas frecuentes desde la infancia, deformidades óseas importantes, baja estatura marcada, escleróticas azuladas o grises, y pérdida de audición. Algunas personas pueden haber tenido decenas o más de 100 fracturas antes de la pubertad en los casos más severos.
- Tipo IV (moderado): gravedad intermedia entre los tipos I y III; fracturas y deformidades óseas de grado variable, escleróticas normales o ligeramente anormales y mayor riesgo de pérdida auditiva.
Además de estos, existen otros tipos (V, VI, VII, etc.) que se definen por hallazgos clínicos, radiológicos y genéticos adicionales. Por ejemplo, algunas variantes presentan calcificaciones o anomalías histológicas específicas.
Síntomas y signos frecuentes
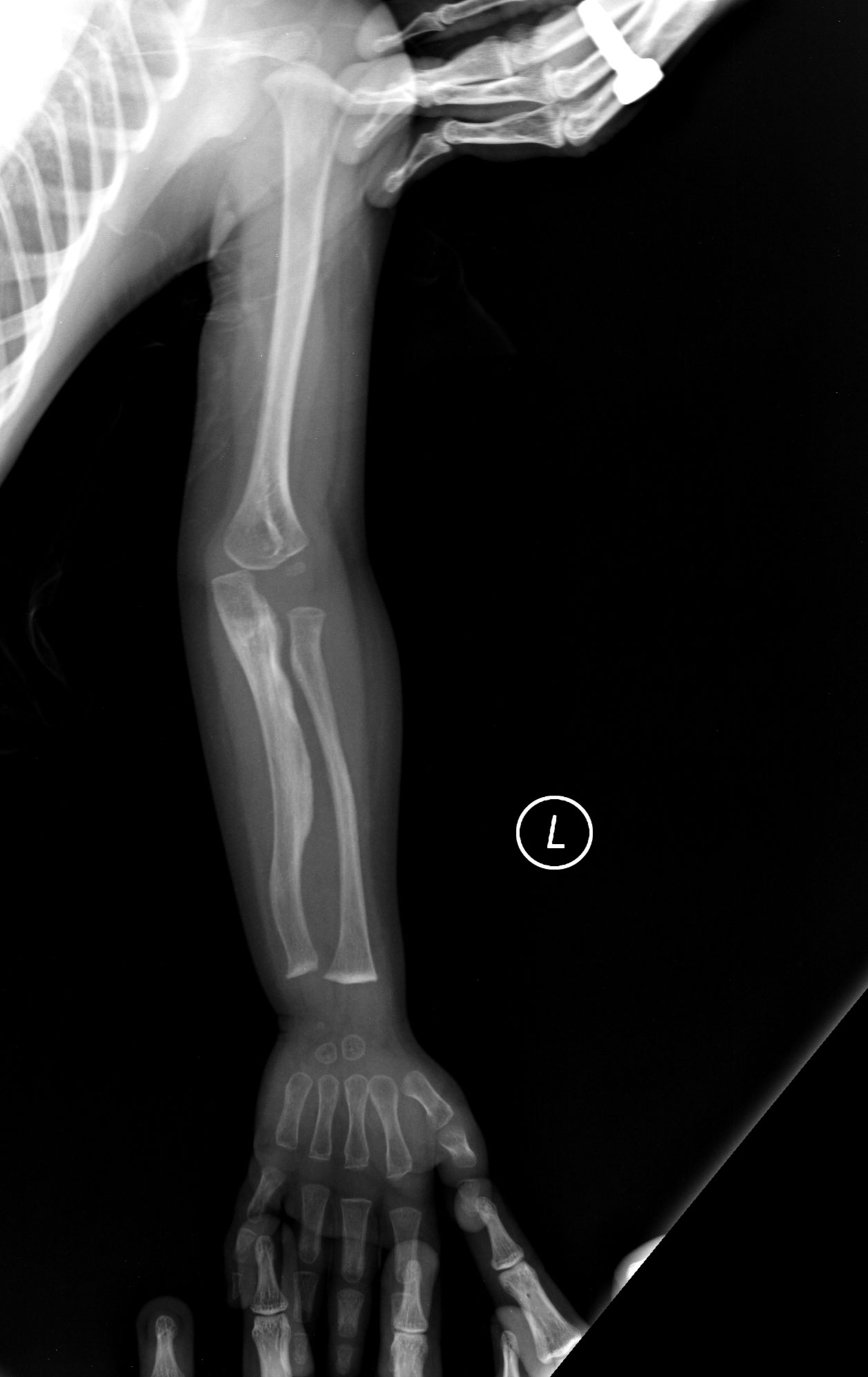
- Fracturas óseas frecuentes, incluso con traumatismos mínimos.
- Osteopenia u osteoporosis infantil, deformidad de huesos largos y columna vertebral (cifosis, escoliosis).
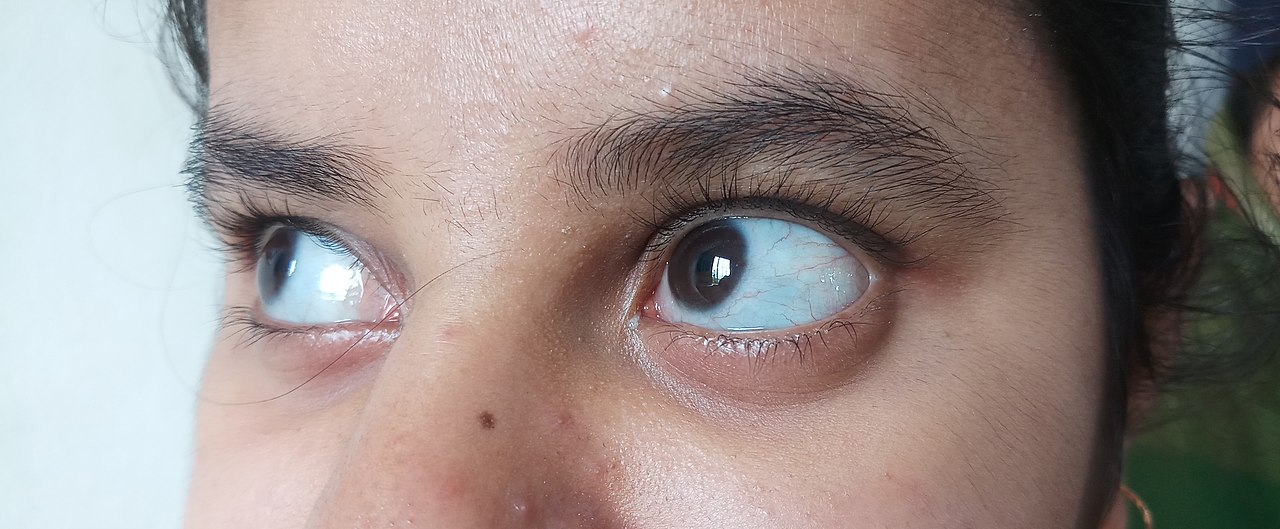
- Escleróticas azuladas, grises o púrpuras (debido a translucidez de la esclera por menor colágeno).
- Dientes frágiles o malformados (dentinogénesis imperfecta).
- Pérdida de audición progresiva (hasta el 50% en la edad adulta en algunos tipos).
- Articulaciones laxas, movilidad articular aumentada y dolores musculoesqueléticos.
- Estatura baja en formas más severas.
- En formas graves, problemas respiratorios por deformidad torácica y fragilidad de costillas.
Diagnóstico
El diagnóstico se basa en la combinación de:
- Historia clínica y familiar (fracturas repetidas, características físicas).
- Examen físico (escleróticas, dentición, deformidades, talla).
- Imágenes radiológicas (fracturas, osteopenia, deformidades óseas).
- Pruebas genéticas para identificar mutaciones en genes asociados (p. ej. COL1A1, COL1A2 y otros).
- En algunos casos, biopsia ósea o análisis bioquímicos del colágeno.
Tratamiento y manejo
No existe una cura definitiva para la OI, pero las intervenciones pueden reducir el número de fracturas, mejorar la función y la calidad de vida. El manejo es multidisciplinario:
- Farmacológico: los bifosfonatos (por ejemplo, pamidronato o ácido zoledrónico) se usan con frecuencia en niños y adultos para aumentar la densidad ósea y disminuir las fracturas. Otros tratamientos experimentales incluyen terapias dirigidas según la alteración genética.
- Quirúrgico: la osteotomía y la colocación de varillas telescópicas intramedulares (rodding) ayudan a corregir deformidades y reducir fracturas recurrentes.
- Rehabilitación: fisioterapia para mejorar la movilidad, la fuerza y prevenir la pérdida de masa muscular; terapia ocupacional para adaptar actividades y reducir el riesgo de caídas.
- Soporte dental: atención odontológica para tratar y prevenir problemas dentales por dentinogénesis imperfecta.
- Audición: evaluación periódica y dispositivos auditivos si hay pérdida auditiva.
- Cuidados respiratorios y cardiológicos: vigilancia en casos con afectación torácica o cardiaca.
- Consejería genética: orientación para la planificación familiar, diagnóstico prenatal o preimplantacional cuando procede.
Pronóstico y calidad de vida
El pronóstico depende del tipo y de la gravedad de la OI. Muchas personas con formas leves llevan una vida activa con adaptaciones y tratamiento adecuado. Las formas más graves pueden requerir cuidados continuos y presentan mayor morbilidad y riesgo vital en la infancia. La detección temprana, el tratamiento integral y el seguimiento multidisciplinario mejoran los resultados y la autonomía.
Consejos prácticos para pacientes y cuidadores
- Crear un entorno doméstico seguro: suelos antideslizantes, buenos apoyos, evitar caídas.
- Control nutricional: aporte suficiente de calcio y vitamina D según indicación médica.
- Actividad física adaptada: ejercicios de bajo impacto para mantener fuerza y movilidad (natación, fisioterapia supervisada).
- Atención ante traumatismos: acudir a valoración médica ante dolor óseo o sospecha de fractura.
- Búsqueda de un equipo médico con experiencia en OI (ortopedia pediátrica/adulta, genética, rehabilitación, odontología y otorrinolaringología).
Si sospecha que usted o un familiar padecen osteogénesis imperfecta, consulte con un equipo médico especializado para evaluación genética y planificación del manejo. La intervención precoz y la atención multidisciplinaria son clave para reducir complicaciones y mejorar la calidad de vida.
Síntomas
Los síntomas menos graves de la OI pueden incluir:
- huesos que se rompen fácilmente
- articulaciones sueltas
- bajo tono muscular
- color azul, púrpura o gris en la parte normalmente blanca de los ojos
- forma triangular de la cara
- tendencia a desarrollar escoliosis
- dientes frágiles
La OI tiene muchos otros síntomas graves y mortales, como problemas respiratorios y deformidad ósea.
Datos demográficos
Preguntas y respuestas
P: ¿Qué es la osteogénesis imperfecta?
R: La osteogénesis imperfecta es un trastorno genético comúnmente denominado enfermedad de los huesos frágiles. Debilita o destruye la varilla de colágeno, que proporciona resistencia a los huesos, y da lugar a huesos más propensos a romperse.
P: ¿Cómo se hereda la osteogénesis imperfecta?
R: La osteogénesis imperfecta suele ser una enfermedad autosómica dominante, lo que significa que una persona puede padecerla si sólo uno de sus progenitores tiene el gen anómalo.
P: ¿Quién identificó por primera vez la osteogénesis imperfecta?
R: Vrolik identificó por primera vez la osteogénesis imperfecta en 1849.
P: ¿Existe cura para la osteogénesis imperfecta?
R: Desgraciadamente, la osteogénesis imperfecta no tiene cura.
P: ¿Cuáles son los cuatro tipos de osteogénesis imperfecta?
R: Los cuatro tipos de osteogénesis imperfecta son el tipo uno, el tipo dos, el tipo tres y el tipo cuatro.
P: ¿Cuáles son los síntomas de la osteogénesis imperfecta de tipo tres?
R: Las personas con el tipo tres de osteogénesis imperfecta pueden tener más de 100 fracturas antes de la pubertad. Sus ojos suelen desarrollar un tinte morado, azul o gris y las personas con este caso también suelen tener pérdida de audición.
P: ¿Es frecuente la pérdida de audición en las personas con osteogénesis imperfecta?
R: El 50% de las personas que padecen osteogénesis imperfecta tienen pérdida auditiva al llegar a la edad adulta.
Artículos relacionados
Autor
AlegsaOnline.com Osteogénesis imperfecta (huesos frágiles): definición, tipos y síntomas Leandro Alegsa
URL: https://es.alegsaonline.com/art/73422
Fuentes
- oif.org : "Osteogenesis Imperfecta Foundation:"
- nlm.nih.gov : "Autosomal dominant: MedlinePlus Medical Encyclopedia"
- oif.org : "Osteogenesis Imperfecta Foundation: Understanding Bone Structure"