Cicloadición: definición, tipos y ejemplos (Diels–Alder, [3+2])
Cicloadición: definición, tipos y ejemplos (Diels–Alder, [3+2]). Explicación clara de mecanismos, ejemplos y aplicaciones de reacciones pericíclicas para estudiantes e investigadores.
Una cicloadición es una reacción química entre reactivos con dobles enlaces que se sustituyen por una estructura de anillo. Es una reacción química pericíclica en la que "dos o más moléculas insaturadas (o partes de la misma molécula) se combinan con la formación de un aducto cíclico en el que hay una reducción neta de la multiplicidad de enlaces". Se trata de una reacción de ciclización: se forma un nuevo anillo de átomos.
Las cicloadiciones se denominan según el tamaño básico de las moléculas que se unen. De este modo, la reacción de Diels-Alder sería una [4 + 2]cicloadición, y la cicloadición 1,3-dipolar una [3 + 2]cicloadición. Este tipo de reacción es una reacción de adición no polar.
Galería de imágenes
6 Imágenes





Clasificación y ejemplos comunes
- [4+2] — Diels–Alder: reacción entre un dieno conjugado (4 electrones π) y un dienófilo (2 electrones π) que forma un ciclohexeno o derivados. Muy utilizada en síntesis orgánica por su alta regiocontrol y estereoselectividad.
- [3+2] — Cicloadición 1,3-dipolar: reacción entre un dipolo 1,3 (por ejemplo, una azida, óxido nitrógeno o nitrógeno y carbono con carga distribuida) y un dipolarófilo (alqueno o alquino) que forma anillos de cinco miembros. Un ejemplo práctico es la reacción azida–alquino (Huisgen), y su versión catalizada por cobre (click chemistry) que produce 1,2,3-triazoles con alta selectividad.
- [2+2]: forma ciclobutanos. La cicloadición [2+2] es térmicamente prohibida (concertada) por las reglas de conservación de la simetría, pero suele ocurrir bajo irradiación (fotoquímica) o por mecanismos en dos pasos (radicales o iónicos).
- Otras combinaciones: [6+4], [5+2] y otras cicloadiciones más exóticas aparecen en síntesis y en química de materiales.
Mecanismo y teoría de orbitales
Muchas cicloadiciones son reacciones concertadas, es decir, los enlaces se forman y se rompen en un único estado de transición sin intermedios detectables. La explicación moderna se basa en la teoría de orbitales fronterizos (FMO): la interacción favorable entre el HOMO de un reactivo y el LUMO del otro dirige la reactividad, la regioconfiguración y la estereoselectividad.
Las reglas de Woodward–Hoffmann (o reglas de conservación de la simetría) predicen qué procesos concertados son termoquímicamente permitidos o prohibidos, teniendo en cuenta si las interacciones son suprafaciales (misma cara) o antarafaciales (caras opuestas). Por ejemplo, la [4+2]Diels–Alder térmica es suprafacial-suprafacial y está permitida, mientras que la [2+2] concertada térmica está prohibida pero permitida bajo irradiación (fotocatalizada) al cambiar la ocupación electrónica.
Estereo- y regioselectividad
- Estereoespecificidad: en muchas cicloadiciones concertadas la configuración de los reactivos se transmite al producto (p. ej., los sustituyentes cis en un dienófilo permanecen cis en el producto del Diels–Alder).
- Regioselectividad: se predice mediante las interacciones FMO (coeficientes orbitalarios) y por efectos electrostáticos. En Diels–Alder existen reglas empíricas (normalmente el diente más nucleofílico se une al más electrofílico) y a menudo se observa la regla del endo (preferencia por el aducto endo cuando hay grupos aceptores de electrones en el dienófilo, debido a interacciones secundarias en el estado de transición).
Condiciones experimentales y catalizadores
Las cicloadiciones se realizan bajo condiciones térmicas o fotoquímicas según el tipo. Para mejorar rendimiento y selectividad se emplean:
- Ácidos de Lewis (BF3, AlCl3, ZnCl2, etc.) que coordinan al dienófilo y disminuyen su LUMO, acelerando reacciones como el Diels–Alder y a menudo favoreciendo la regla del endo.
- Catalizadores quirales para inducir enantioselectividad en respuestas asimétricas (por ejemplo catalizadores metálicos o organocatalizadores quirales).
- Fotocatálisis para permitir cicloadiciones prohibidas térmicamente (p. ej., [2+2]) o para activar componentes mediante transferencia de energía o electrones.
Mecanismos stepwise y excepciones
No todas las cicloadiciones son estrictamente concertadas: algunas proceden por vías en dos pasos con intermedios radicálicos o iónicos (por ejemplo, ciertas [2+2] térmicas o reacciones en presencia de radicales). La distinción experimental entre mecanismo concertado y paso a paso se realiza mediante estudios cinéticos, isotópicos, y detección de intermedios.
Aplicaciones
- Síntesis de productos naturales y de fármacos: formación eficiente y estereocontrolada de anillos de 5 y 6 miembros.
- Química “click”: la cicloadición 1,3-dipolar azida–alquino (versión catalizada por Cu(I)) es una herramienta valiosa en bioconjugación, materiales y química medicinal.
- Polimerización y materiales: cicloadiciones controladas se usan para obtener redes, monómeros cíclicos y polímeros con propiedades específicas.
Consideraciones prácticas
- Planificar sustituyentes para dirigir regio- y estereoselectividad mediante efectos electrónicos y estéricos.
- Usar catalizadores adecuados (ácidos de Lewis o catalizadores quirales) para mejorar rendimiento y enantioselectividad.
- Evitar condiciones oxidantes o fuentes de radicales si se desea mantener un mecanismo concertado.
Resumen
La cicloadición es una clase central de reacciones en química orgánica que permite construir anillos de manera eficiente y con alto control estereo/regioquímico. Comprender la teoría de orbitales, las reglas de simetría y las condiciones experimentales permite diseñar rutas sintéticas exitosas: desde la clásica reacción de Diels-Alder ([4+2]) hasta las cicloadiciones 1,3-dipolares ([3+2]) y las variantes fotoquímicas ([2+2]).
Mecanismo de reacción
El calor puede hacer que los dobles enlaces formen un anillo. Las cicloadiciones térmicas suelen tener (4n + 2) electrones π participando en el material de partida, para algún número entero n. Debido a la simetría orbital, la mayoría de las cicloadiciones son suprafaciales-suprafaciales. En raras ocasiones, son antarafaciales-antarafaciales. Hay algunos ejemplos de cicloadiciones térmicas que tienen 4n π electrones (por ejemplo la cicloadición [2 + 2]). Estas proceden en un sentido suprafacial-antarafacial. Por ejemplo, la dimerización del ceteno tiene un conjunto ortogonal de orbitales p. Esos orbitales p permiten que la reacción proceda utilizando un estado de transición cruzado.
La luz también puede hacer que los dobles enlaces formen un anillo. Las cicloadiciones en las que participan 4n electrones π también pueden producirse como resultado de la activación fotoquímica. Aquí, un componente hace que un electrón se mueva del orbital molecular más ocupado (HOMO) (enlace π) al orbital molecular más bajo desocupado (LUMO) (antienlace π*). Después de que el electrón es promovido al orbital más alto, la simetría orbital permite que la reacción proceda de manera suprafacial-suprafacial. Un ejemplo es la reacción de DeMayo. Otro ejemplo es la dimerización fotoquímica del ácido cinámico.
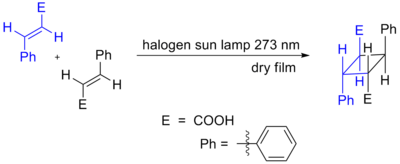
Nótese que no todas las ciclizaciones fotoquímicas (2+2) son cicloadiciones; se sabe que algunas operan por mecanismos radicales.
Algunas cicloadiciones en lugar de enlaces π operan a través de anillos de ciclopropano tensados; ya que éstos tienen un carácter π significativo. Por ejemplo, un análogo de la reacción de Diels-Alder es la reacción de cuadriciclo-DMAD:
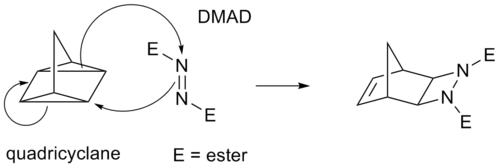
En la notación de (i+j+...) de la cicloadición, i y j se refieren al número de átomos que participan en la cicloadición. En esta notación una reacción de Diels-Alder es una (4+2)cicloadición y una adición 1,3-dipolar como el primer paso en la ozonólisis es una (3+2)cicloadición. Esta notación utiliza paréntesis. Sin embargo, la notación preferida por la IUPAC, con [i+j+...] cuenta los electrones y no los átomos. Utiliza corchetes. En esta notación, la reacción de Diels-Alder y la reacción dipolar se convierten en una [4+2]cicloadición. La reacción entre el norbornadieno y un alquino activado es una [2+2+2]cicloadición.
Tipos de cicloadición
Reacciones de Diels-Alder
La reacción de Diels-Alder es una reacción de [4+2]cicloadición.
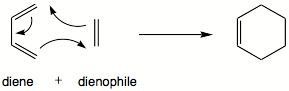
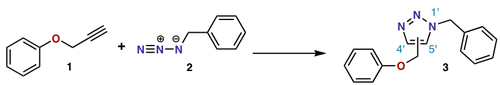
Cicloadiciones de Huisgen
La reacción de cicloadición de Huisgen es una cicloadición [2+3].
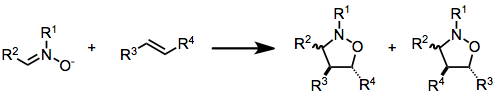
Cicloadición nitrona-olefina
La cicloadición nitrona-olefina es una cicloadición [3+2].
Cicloadiciones formales
Las cicloadiciones tienen a menudo análogos catalizados por metales y radicales escalonados, pero no son reacciones pericíclicas en sentido estricto. Cuando en una cicloadición intervienen intermediarios cargados o radicales, o cuando el resultado de la cicloadición se encuentra en una serie de pasos de reacción, a veces se denominan cicloadiciones formales para diferenciarlas de las verdaderas cicloadiciones pericíclicas.
Un ejemplo de una cicloadición formal [3+3]entre una enona cíclica y una enamina catalizada por el n-butilitio es una reacción en cascada de enamina / 1,2-adición de Stork:
![Intermolecular Formal [3+3] Cycloaddition Reaction](https://www.alegsaonline.com/image/600px-3%2B3-cycloaddition.svg.png)
Preguntas y respuestas
P: ¿Qué es una cicloadición?
R: Una cicloadición es una reacción química entre reactivos con dobles enlaces que se sustituyen por una estructura de anillo.
P: ¿Qué tipo de reacción química es una cicloadición?
R: Una cicloadición es una reacción química pericíclica en la que "dos o más moléculas insaturadas (o partes de la misma molécula) se combinan con la formación de un aducto cíclico en el que se produce una reducción neta de la multiplicidad de enlaces."
P: ¿Qué hace una reacción de cicloadición?
R: Una cicloadición es una reacción de ciclización: forma nuevos anillos de átomos.
P: ¿Cómo se denominan las cicloadiciones?
R: Las cicloadiciones se denominan según el tamaño básico de las moléculas que se unen.
P: ¿Qué es la reacción de Diels-Alder?
R: La reacción de Diels-Alder es una cicloadición [4 + 2].
P: ¿Qué es la cicloadición 1,3-dipolar?
R: La cicloadición 1,3-dipolar es una cicloadición [3 + 2].
P: ¿Qué tipo de reacción es una cicloadición?
R: Una cicloadición es una reacción de adición no polar.
Artículos relacionados
Autor
AlegsaOnline.com Cicloadición: definición, tipos y ejemplos (Diels–Alder, [3+2]) Leandro Alegsa
URL: https://es.alegsaonline.com/art/24866
Fuentes
- goldbook.iupac.org : Cycloaddition
- doi.org : 10.1021/ed083p940
- doi.org : 10.1002/anie.200603302
- pubmed.ncbi.nlm.nih.gov : 17146819