Química computacional: definición, métodos y aplicaciones en diseño de fármacos
Química computacional: métodos, aplicaciones y su papel en el diseño de fármacos. Descubre cómo simulan moléculas, predicen propiedades y aceleran la investigación farmacéutica.
La química computacional es una rama de la química que utiliza la informática para ayudar a resolver problemas químicos. Estos programas calculan las estructuras y propiedades de las moléculas y los sólidos. La química computacional suele complementar la información obtenida mediante experimentos químicos. Puede predecir fenómenos químicos que aún no se han observado. Se utiliza mucho en el diseño de nuevos fármacos y materiales.
La química computacional puede predecir la estructura (es decir, las posiciones esperadas de los átomos de la molécula), las energías absolutas y relativas (de interacción), las distribuciones de carga electrónica, los dipolos y los momentos multipolares superiores, las frecuencias vibracionales, la reactividad u otras magnitudes espectroscópicas y las secciones transversales de colisión con otras partículas.
La química computacional estudia sistemas tanto estáticos como dinámicos. En todos los casos, a medida que aumenta el tamaño del sistema estudiado, también crece el tiempo de computación y otros recursos (como la memoria y el espacio en disco) utilizados. Ese sistema puede ser una sola molécula, un grupo de moléculas o un sólido. Los métodos de la química computacional van desde los muy precisos hasta los muy aproximados. Los métodos de alta precisión suelen ser factibles sólo para sistemas pequeños.
Galería de imágenes
8 Imágenes


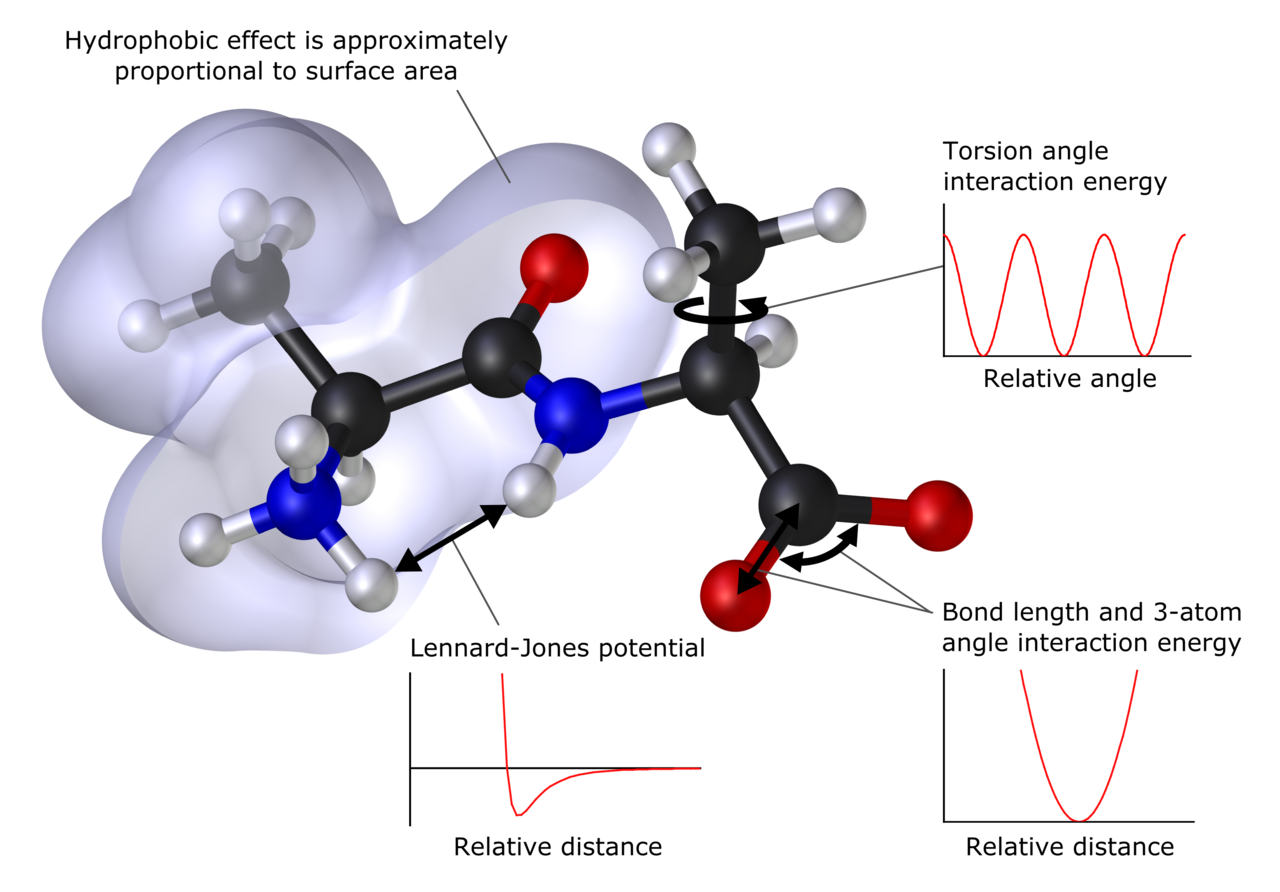


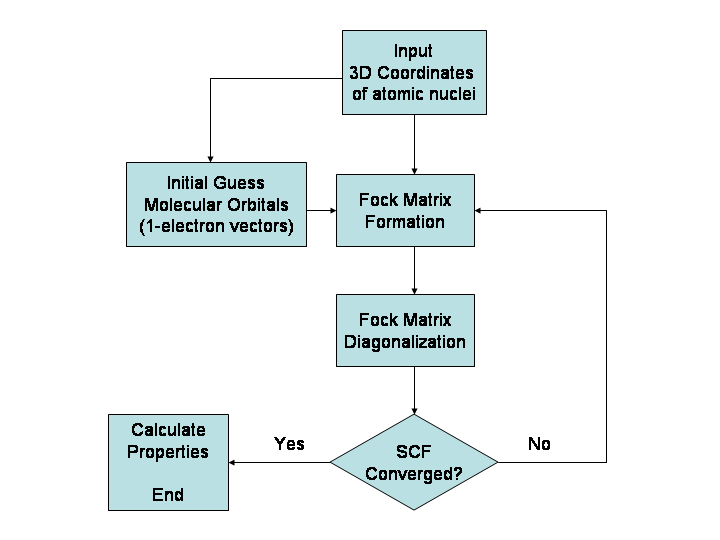
Métodos principales
- Mecánica cuántica (QM): calcula propiedades electrónicas desde primeros principios. Incluye:
- Ab initio: Hartree–Fock (HF) y métodos correlacionados (MP2, CCSD(T)). Muy precisos para sistemas pequeños pero costosos.
- Teoría del funcional de la densidad (DFT): buen balance entre coste y precisión para geometrías, energías y espectros; depende de la elección del funcional.
- Métodos semiempíricos (AM1, PMx): aproximaciones parametrizadas que reducen costes y permiten estudiar sistemas más grandes con menor precisión que DFT.
- Mecánica molecular (MM) y campos de fuerza: tratan átomos como partículas con potenciales clásicos (bondes, ángulos, torsiones, no enlazadas). Adecuados para macromoléculas (proteínas, lípidos, polímeros). Ejemplos de campos: AMBER, CHARMM, OPLS, MMFF.
- Dinámica molecular (MD): simula la evolución temporal de un sistema usando MM o QM/MM. Permite estudiar flexibilidad, plegamiento, difusión y procesos dependientes del tiempo.
- QM/MM: métodos híbridos que combinan QM para la región reactiva y MM para el entorno, útiles en catálisis enzimática y reacciones en solución.
- Monte Carlo (MC): técnica de muestreo estadístico usada para estudiar equilibrio termodinámico, configuraciones conformacionales y propiedades termodinámicas.
- Docking y cribado virtual: predicen poses de unión y afinidades relativas de ligandos frente a dianas macromoleculares; se usan en el diseño y selección inicial de compuestos.
- Métodos para energías libres: MM/PBSA, MM/GBSA, integración termodinámica (TI), y free energy perturbation (FEP) calculan diferencias de energía libre de unión con mayor riguroso teórico (y mayor coste).
- Modelado basado en ligando: QSAR, farmacóforos y modelos de predicción de actividad que correlacionan estructuras químicas con propiedades biológicas.
- Aprendizaje automático y redes neuronales: modelos para predecir propiedades, generar nuevas moléculas y acelerar potenciales empíricos (potenciales neuronales).
Aplicaciones en diseño de fármacos
- Identificación y validación de dianas: modelado de la estructura de proteínas (homología, modelado por IA), predicción de sitios activos y análisis de pockets.
- Cribado virtual y docking: selección de hits entre bibliotecas virtuales mediante docking y filtrado por propiedades físicoquímicas.
- Optimización de leads: análisis SAR asistido por cálculo de energías, interacción proteína–ligando, y predicción de afinidad por métodos de energías libres.
- Modelado de mecanismos de unión: MD y QM/MM para estudiar rutas de unión, cambios conformacionales inducidos y contribuciones entálpicas/entrópicas.
- Predicción ADMET: modelos computacionales para absorción, distribución, metabolismo, excreción y toxicidad que ayudan a priorizar compuestos antes de ensayos experimentales.
- Diseño de novo y fragment-based design: herramientas que ensamblan o generan moléculas con criterios de afinidad y sintetizabilidad.
- Apoyo a síntesis y química médica: predicción de selectividad, rutas reactivas y estabilidad química.
Otras áreas de aplicación
- Materiales y nanociencia: diseño de materiales con propiedades electrónicas, ópticas o mecánicas específicas.
- Catalisis: estudio de mecanismos, intermediarios y barreras de reacción para diseñar mejores catalizadores.
- Espectroscopía teórica: predicción de espectros IR, Raman, NMR, UV-Vis y cálculo de propiedades observables para asignar señales experimentales.
- Química atmosférica y colisiones: modelado de procesos de colisión y reacciones en fase gas.
Limitaciones y buenas prácticas
- Escalado computacional: los métodos más precisos son prohibitivos para sistemas grandes; suele usarse una jerarquía de técnicas (p. ej. DFT para fragmentos, MM/MD para entorno macromolecular).
- Precisión y parametrización: los resultados dependen de la elección de funcional, base, campo de fuerza y parámetros; la validación contra datos experimentales y benchmarks es esencial.
- Solvatación y entropía: efectos de solvente y contribuciones entrópicas son críticos para la predicción de afinidades y deben tratarse con modelos explícitos o implícitos y muestreos adecuados.
- Muestreo conformacional: asegurar un muestreo suficiente (simulaciones largas, réplicas, métodos de muestreo mejorado) es clave para resultados fiables.
- Reproducibilidad: documentar condiciones (temperatura, presión, parámetros, versiones de software) y, cuando sea posible, compartir scripts y datos de entrada/salida.
Recursos y tendencias actuales
- Herramientas y software: existen paquetes especializados para QM (Gaussian, ORCA, NWChem, GAMESS), para MD (GROMACS, AMBER, CHARMM, NAMD) y suites integradas (Schrödinger, OpenMM, AutoDock). El uso de GPUs ha acelerado enormemente MD y algunos métodos DFT/MD.
- Aprendizaje automático: modelos de IA para generar moléculas, predecir propiedades y construir potenciales de alta precisión están transformando flujos de trabajo de descubrimiento.
- Computación cuántica y exascale: investigación activa para aprovechar nuevas arquitecturas en problemas cuánticos y en la simulación de sistemas químicos difíciles para métodos clásicos.
Consejos prácticos para proyectos de diseño de fármacos
- Elegir el método adecuado según la pregunta: precisión vs coste computacional.
- Combinar técnicas: por ejemplo, docking para cribado inicial, MD para muestreo conformacional y FEP/TI para estimaciones cuantitativas de afinidad.
- Validar con datos experimentales siempre que sea posible y usar controles/benchmarks.
- Documentar y versionar datos y parámetros para garantizar reproducibilidad.
En resumen, la química computacional es una disciplina diversa que ofrece herramientas poderosas para entender y predecir fenómenos químicos y acelerar el diseño de fármacos y materiales. Su eficacia depende de la selección adecuada de métodos, del tratamiento riguroso del muestreo y solvatación, y de la validación sistemática frente a experimentos.

Páginas relacionadas
- Bioinformática
- Mecánica estadística
Preguntas y respuestas
P: ¿Qué es la química computacional?
R: La química computacional es una rama de la química que utiliza la informática para ayudar a resolver problemas químicos. Puede utilizarse para calcular las estructuras y propiedades de moléculas y sólidos, predecir fenómenos químicos que aún no se han observado y diseñar nuevos fármacos y materiales.
P: ¿Qué tipos de sistemas estudia la química computacional?
R: La química computacional estudia tanto sistemas estáticos como dinámicos. El sistema puede ser una única molécula, un grupo de moléculas o un sólido.
P: ¿Qué tipos de información puede proporcionar la química computacional?
R: La química computacional puede proporcionar información como la estructura (posiciones de los átomos), energías absolutas y relativas, distribuciones de carga electrónica, dipolos y momentos multipolares superiores, frecuencias vibracionales, reactividad u otras magnitudes espectroscópicas, y secciones transversales de colisión con otras partículas.
P: ¿Cuál es la precisión de los métodos utilizados en química computacional?
R: La precisión de los métodos utilizados en química computacional varía de muy precisa a muy aproximada. Los métodos altamente precisos suelen ser factibles sólo para sistemas pequeños.
P: ¿Cómo complementa la química computacional los datos experimentales?
R: La química computacional suele complementar la información obtenida mediante experimentos químicos. Puede utilizarse para predecir resultados que aún no se han observado experimentalmente.
P: ¿Afecta el tamaño del sistema estudiado a la cantidad de tiempo que se necesita en el ordenador?
R: Sí. A medida que aumenta el tamaño del sistema estudiado, también lo hace la cantidad de tiempo de ordenador necesario para el análisis, así como recursos como la memoria y el espacio en disco necesarios para el almacenamiento.
Artículos relacionados
Autor
AlegsaOnline.com Química computacional: definición, métodos y aplicaciones en diseño de fármacos Leandro Alegsa
URL: https://es.alegsaonline.com/art/22297