Síndromes progeroides: qué son, causas genéticas y ejemplos (HGPS, Werner)
Síndromes progeroides: descubre qué son, sus causas genéticas y ejemplos clave (HGPS, Werner). Causas, síntomas y avances en investigación sobre envejecimiento acelerado.
Los síndromes progeroides (SP) describen una serie de trastornos genéticos en los que la persona afectada parece envejecer más rápido. Todos estos trastornos son monogenéticos, es decir, proceden de mutaciones de un solo gen. La mayoría de las mutaciones conocidas de los PS conducen a defectos en el mecanismo de reparación del ADN o a defectos en una proteína conocida como lamin A/C.
Progeroide significa "parecido a la vejez". Esta definición puede aplicarse a muchas enfermedades diferentes. La enfermedad de Alzheimer y la de Parkinson sólo afectan a un tejido. En la mayoría de los casos, el término síndrome progeroide se utiliza para los casos en los que las personas afectadas sólo muestran algunos de los rasgos del envejecimiento, pero no todos. En estos casos, se ven afectados muchos tipos de tejidos diferentes.
Los individuos con trastornos relacionados con la progeria suelen tener una esperanza de vida reducida. Los síndromes progeroides más estudiados son el síndrome de Werner (WS) y el síndrome de progeriade Hutchinson-Gilford(HGPS), porque se parecen al envejecimiento natural.
Debido a su propiedad de acelerar el envejecimiento (senescencia), los síndromes progeroides han sido ampliamente estudiados en los campos del envejecimiento, la regeneración, las células madre y el cáncer.
Galería de imágenes
3 Imágenes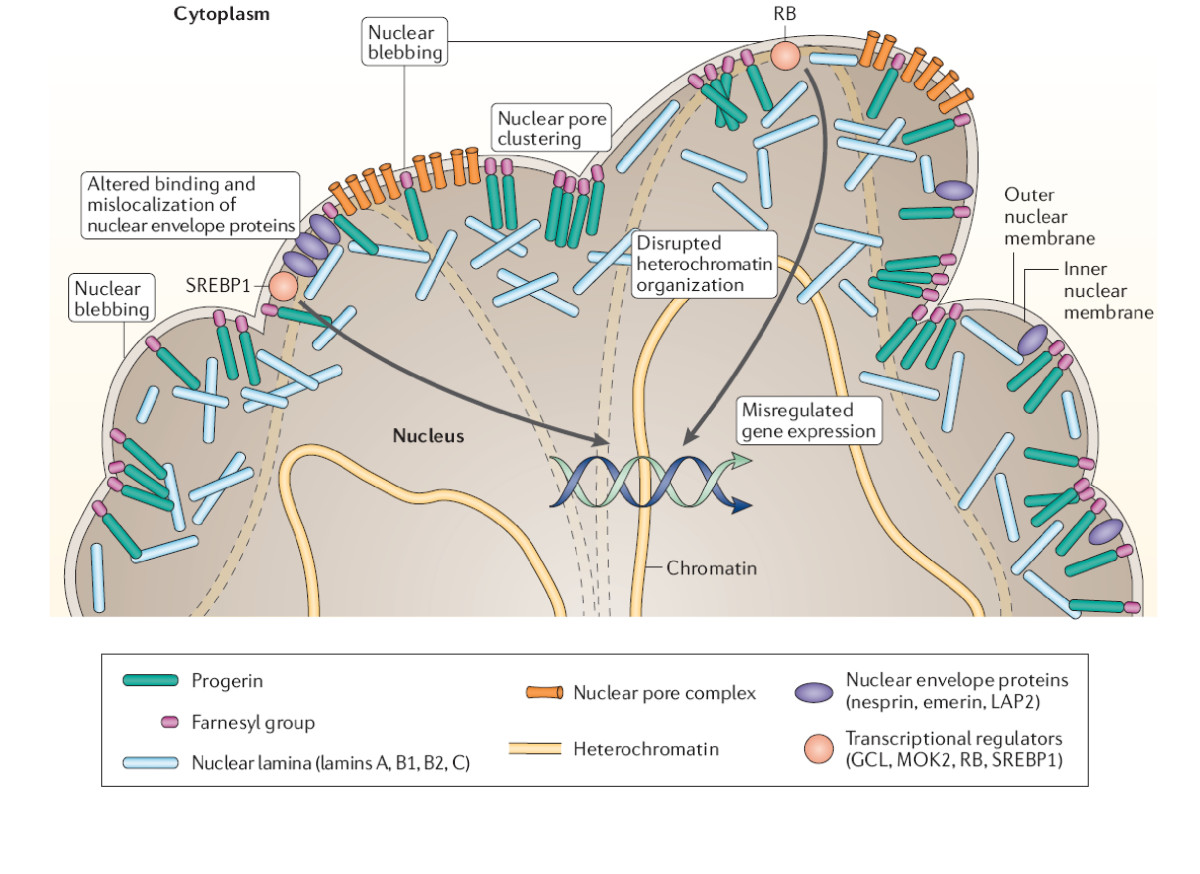

Causas genéticas y mecanismos
Aunque los síndromes progeroides son heterogéneos, pueden agruparse según los mecanismos moleculares principales:
- Defectos en reparación del ADN: Mutaciones en genes implicados en la reparación del ADN (por ejemplo, WRN, BLM, genes de la reparación por escisión de nucleótidos) provocan acumulación de daño genómico, inestabilidad cromosómica y aceleración de procesos relacionados con el envejecimiento celular.
- Alteraciones de la lámina nuclear: Mutaciones en el gen LMNA (que codifica lamin A/C) o en genes implicados en el procesamiento de laminas (por ejemplo, ZMPSTE24) generan una proteína anómala llamada progerina o deficiencias estructurales de la envoltura nuclear. Estos defectos alteran la arquitectura nuclear, la expresión génica y la capacidad de la célula para dividirse y repararse.
- Otros mecanismos: Mutaciones en helicasas de la familia RecQ (p. ej. RECQL4), genes implicados en el mantenimiento de la estabilidad genómica o en la reparación del daño por radiación ultravioleta pueden producir fenotipos progeroides parciales o segmentarios.
Cómo se manifiestan (síntomas habituales)
La presentación clínica varía según el síndrome y la edad de inicio, pero los rasgos más frecuentes incluyen:
- Retraso del crecimiento y baja estatura
- Pérdida de cabello (alopecia), canicie prematura
- Pérdida de grasa subcutánea y piel fina o atrófica
- Arrugas faciales y apariencia envejecida
- Rigidez articular, contracturas
- Enfermedad cardiovascular prematura (aterosclerosis, infarto, accidente cerebrovascular)
- Mayor susceptibilidad a ciertos tipos de cáncer (según el síndrome)
- Anomalías oculares (p. ej. cataratas precoces en el síndrome de Werner)
- Problemas metabólicos como diabetes o dislipidemia
Ejemplos principales
- Síndrome de Hutchinson‑Gilford (HGPS): Generalmente causado por una mutación puntual en LMNA (la variante más frecuente es la c.1824C>T, p.G608G) que activa un sitio de empalme aberrante y produce progerina, una forma truncada de lamin A. Se manifiesta en la infancia con crecimiento lento, pérdida de cabello, piel rígida, pérdida de grasa subcutánea y enfermedad cardiovascular prematura. La supervivencia media suele situarse en la adolescencia temprana; la causa más común de muerte son las complicaciones cardiovasculares.
- Síndrome de Werner (WS): Producido por mutaciones en el gen WRN, que codifica una helicasa de la familia RecQ. A diferencia del HGPS, el WS suele iniciarse en la adolescencia tardía o adultez joven y muestra canicie y calvicie prematuras, cataratas bilaterales, ulceraciones cutáneas, calcificaciones y alto riesgo de aterosclerosis, diabetes y varios tipos de cáncer. La expectativa de vida se reduce, frecuentemente hasta mediados de la vida adulta.
- Otros síndromes relacionados: Bloom (mutación en BLM), Cockayne (mutaciones en ERCC6/ERCC8), xeroderma pigmentoso (defectos en la reparación por escisión de nucleótidos), Rothmund‑Thomson (RECQL4), mandibuloacral dysplasia (mutaciones en LMNA o ZMPSTE24), entre otros. Cada uno muestra un perfil clínico y riesgos distintos, y algunos son segmentarios (afectan tejidos concretos) en lugar de producir un envejecimiento global.
Diagnóstico
- Evaluación clínica basada en los signos característicos (edad de comienzo, patrón de afectación cutánea, crecimiento, cataratas, cardiopatía, etc.).
- Confirmación genética mediante secuenciación del/los genes sospechosos (p. ej. LMNA para HGPS, WRN para Werner). Pruebas de paneles genéticos o exoma cuando el diagnóstico no es claro.
- Pruebas complementarias: análisis cardiológicos (ecocardiograma, calcificación coronaria), evaluación endocrinometabólica, estudios oncológicos según el riesgo del síndrome.
Manejo y pronóstico
No existe una cura universal para los síndromes progeroides. El tratamiento es mayoritariamente sintomático y de prevención de complicaciones:
- Vigilancia cardiovascular intensiva y control de factores de riesgo (hipertensión, lípidos, diabetes).
- Tratamiento de complicaciones: cirugía de cataratas, cuidado de úlceras y problemas cutáneos, manejo ortopédico de contracturas.
- Screening oncológico adaptado al riesgo específico de cada síndrome.
- Soporte multidisciplinar: dermatología, cardiología, endocrinología, genética clínica, fisioterapia y apoyo psicosocial.
- Consejería genética para familias: explicando el riesgo de recurrencia, opciones de diagnóstico prenatal o diagnóstico genético preimplantacional cuando procede.
Investigación y terapias experimentales
Las investigaciones han identificado dianas terapéuticas prometedoras, especialmente en HGPS:
- Inhibidores de la farnesiltransferasa (FTI): fármacos que reducen la interacción nociva de progerina con la membrana nuclear; lonafarnib ha mostrado beneficios clínicos y reducción de mortalidad en estudios de HGPS.
- Otras aproximaciones moleculares: oligonucleótidos antisentido para modificar el empalme de LMNA, compuestos que favorecen la eliminación de progerina (p. ej. rapamicina en estudios preclínicos) y terapias basadas en edición génica en investigación preclínica.
- Ensayos clínicos y modelos celulares/animales: continúan explorando combinaciones de fármacos y estrategias para restaurar la función de la lámina nuclear o mejorar la reparación del ADN.
Consideraciones finales
Los síndromes progeroides ofrecen una ventana única para estudiar los mecanismos del envejecimiento humano y para desarrollar terapias dirigidas. Aunque son enfermedades raras, los avances en genética, medicina molecular y ensayos clínicos han empezado a traducirse en tratamientos que mejoran la calidad y duración de la vida en algunos casos. El manejo óptimo requiere un enfoque multidisciplinar y el seguimiento homogéneo de complicaciones cardiometabólicas y oncológicas.
Preguntas y respuestas
P: ¿Qué son los síndromes progeroides (SP)?
R: Los síndromes progeroides son una serie de trastornos genéticos en los que la persona afectada parece envejecer más rápido.
P: ¿Qué causa los PS?
R: La mayoría de las mutaciones conocidas de los PS provocan defectos en el mecanismo de reparación del ADN o defectos en una proteína conocida como lamin A/C.
P: ¿Qué significa progeroide?
R: Progeroide significa parecido a la vejez.
P: ¿Las enfermedades de Alzheimer y Parkinson pueden considerarse síndromes progeroides?
R: No, las enfermedades de Alzheimer y Parkinson sólo afectan a un tejido, y el término síndrome progeroide se utiliza para los casos en los que las personas afectadas sólo muestran algunos de los rasgos del envejecimiento, pero no todos.
P: ¿Cuántos tipos diferentes de tejidos pueden verse afectados en los síndromes progeroides?
R: En los síndromes progeroides se ven afectados muchos tipos diferentes de tejidos.
P: ¿Cuál es la esperanza de vida de los individuos con trastornos relacionados con la PS?
R: Los individuos con trastornos relacionados con la PS suelen tener una esperanza de vida reducida.
P: ¿Cuáles son los síndromes progeroides más estudiados y por qué?
R: Los síndromes progeroides más estudiados son el síndrome de Werner (SW) y el síndrome de progeria de Hutchinson-Gilford (HGPS), porque se asemejan al envejecimiento natural.
Artículos relacionados
Autor
AlegsaOnline.com Síndromes progeroides: qué son, causas genéticas y ejemplos (HGPS, Werner) Leandro Alegsa
URL: https://es.alegsaonline.com/art/79367
Fuentes
- doi.org : 10.1093/hmg/ddl214
- pubmed.ncbi.nlm.nih.gov : 16987878