Acromegalia: qué es, causas, síntomas, diagnóstico y tratamiento
Acromegalia: descubre causas, síntomas, diagnóstico y opciones de tratamiento para evitar complicaciones. Guía clara para detectar y tratar a tiempo.
La acromegalia es un trastorno hormonal poco frecuente que ocurre cuando la glándula pituitaria anterior (adenohipófisis) secreta en exceso la hormona del crecimiento (GH) en la edad adulta. Si el exceso de GH comienza antes de la pubertad, se produce una entidad distinta llamada gigantismo, caracterizada por un crecimiento excesivo en altura. La causa más frecuente de acromegalia es un tumor benigno de la hipófisis denominado "adenoma hipofisario", aunque existen otras causas menos habituales (tumores extrapituitarios que producen GH o GHRH, síndromes genéticos, entre otros).
Galería de imágenes
8 Imágenes






Causas
- Adenoma hipofisario productor de GH: representa la mayoría de los casos. Son generalmente tumores benignos (adenomas) de la adenohipófisis.
- Tumores extrapituitarios: raramente, tumores en el páncreas, pulmón u otros tejidos pueden producir factor liberador de GH (GHRH) o directamente GH.
- Factores genéticos y síndromes raros: mutaciones hereditarias o síndromes como MEN1, McCune–Albright o AIP pueden predisponer a adenomas.
Síntomas y signos
Los síntomas suelen aparecer de forma lenta y progresiva, por eso el diagnóstico a menudo se retrasa (frecuentemente 10–12 años desde el inicio). Entre los hallazgos más frecuentes están:
- Cambios faciales: engrosamiento de labios y nariz, prognatismo (mandíbula prominente), separación de los dientes.
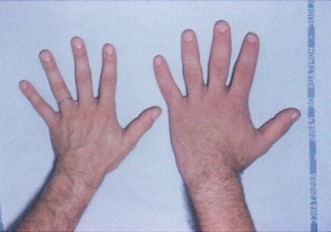
- Agrandamiento de extremidades: aumento del tamaño de manos y pies, anillos y calzado que dejan de servir.
- Problemas músculo-esqueléticos: dolor articular, artropatía degenerativa, síndrome del túnel carpiano.
- Alteraciones metabólicas: resistencia a la insulina, diabetes mellitus, intolerancia a la glucosa.
- Signos cutáneos: sudoración excesiva, piel grasa, aparición de acrocordones.
- Síntomas neurológicos: cefalea y pérdida visual por compresión del quiasma óptico si el adenoma es grande.
- Trastornos sexuales y reproductivos: disminución de la libido, disfunción eréctil, alteraciones menstruales.
- Apnea del sueño: aumento de tejido blando faríngeo favorece episodios de apnea obstructiva.
Diagnóstico
- Pruebas de laboratorio:
- Medición de IGF‑1 (factor de crecimiento similar a la insulina tipo 1): prueba de cribado sensible para exceso crónico de GH.
- Prueba de tolerancia oral a la glucosa (OGTT): en condiciones normales la glucosa suprime la GH; en acromegalia la supresión está ausente o incompleta.
- Evaluación de otras hormonas hipofisarias para detectar hipopituitarismo asociado.
- Imágenes: resonancia magnética (RM) de la silla turca con contraste para localizar y caracterizar el adenoma; la tomografía computarizada (TC) es alternativa si la RM no es posible.
- Evaluación oftalmológica: prueba de campo visual si existe tumor de gran tamaño o síntomas visuales.
- Valoración cardiometabólica: electrocardiograma, ecocardiograma y pruebas metabólicas para detectar complicaciones.
Tratamiento
El objetivo del tratamiento es normalizar los niveles de GH/IGF‑1, reducir o extirpar el tumor y mejorar los síntomas y las comorbilidades. Las opciones principales son:
- Cirugía: la resección transesfenoidal del adenoma hipofisario suele ser la primera opción para la mayoría de los pacientes, especialmente si hay compresión de estructuras adyacentes (por ejemplo, el quiasma óptico). La cirugía puede lograr la curación en adenomas pequeños bien localizados.
- Terapia médica:
- Analogos de somatostatina (octreótido, lanreótido): reducen la secreción de GH y suelen disminuir el tamaño tumoral en muchos casos.
- Pegvisomant (antagonista del receptor de GH): normaliza IGF‑1 bloqueando la acción de la GH, útil cuando los análogos de somatostatina no controlan la enfermedad.
- Dopaminérgicos (cabergolina): a veces eficaz en adenomas con secreción mixta o como terapia adyuvante en casos leves.
- Radioterapia: fraccionada o en forma de radiocirugía estereotáctica (Gamma Knife) en tumores residuales o recurrentes cuando cirugía y fármacos son insuficientes. Su efecto es más lento y puede causar hipopituitarismo a largo plazo.
Complicaciones y pronóstico
Si no se trata, la acromegalia aumenta la morbilidad y la mortalidad, principalmente por enfermedades cardiovasculares, respiratorias y metabólicas. Entre las complicaciones destacadas están:
- Cardiopatía (hipertrofia ventricular, insuficiencia cardiaca).
- Hipertensión arterial y diabetes mellitus.
- Apnea obstructiva del sueño.
- Artropatía e incapacidad funcional por daño osteoarticular.
- Aumento del riesgo de pólipos colorrectales (se recomienda colonoscopia según pautas).
Con un diagnóstico precoz y tratamiento adecuado, la expectativa de vida y la calidad de vida pueden mejorar significativamente.
Seguimiento y objetivos terapéuticos
- Control periódico de IGF‑1 y GH para evaluar la respuesta al tratamiento.
- Imágenes de control (RM) para monitorizar el tamaño tumoral después de cirugía o durante terapia médica.
- Evaluación y tratamiento de comorbilidades (diabetes, hipertensión, apnea, problemas articulares).
- Valoración endocrinológica de por vida: riesgo de recidiva y posible insuficiencia hipofisaria tras cirugía o radioterapia.
Recomendaciones generales
- Si observa cambios progresivos en cara, manos o pies, o desarrolla síntomas metabólicos o respiratorios, consulte con su médico para valoración endocrinológica.
- El tratamiento multidisciplinario (endocrinología, neurocirugía, oftalmología, cardiología, neumología) suele ser necesario para un manejo integral.
- La adherencia al seguimiento y a la terapia médica mejora los resultados a largo plazo.
La acromegalia es una enfermedad tratable; un diagnóstico y un manejo oportunos reducen las complicaciones y mejoran la calidad de vida. Si necesita información más específica sobre pruebas o tratamientos disponibles en su área, consulte con un especialista en endocrinología.
Signos y síntomas
- Dolores de cabeza
- Problemas de visión
- Hirsutismo (en las mujeres): crecimiento del vello en la cara.
- Protuberancia frontal: el hueso de debajo de la frente se hace más grande de lo normal.
- Prognatismo: una parte de la cara sobresale más de lo normal, normalmente la mandíbula inferior.
- Fatiga: la persona suele estar cansada.
- Presión arterial alta
- Papeles de la piel: Pequeño crecimiento en forma de verruga en la piel que no es canceroso.
- Hiperhidrosis: cuando una persona suda demasiado.
- Bromhidrosis: Olor corporal
- Hepatomegalia
- Agrandamiento de manos y pies
- Cardiomiopatía
- Cáncer de colon
Preguntas y respuestas
P: ¿Qué es la acromegalia?
R: La acromegalia es una afección médica que se produce cuando la glándula pituitaria produce demasiada hormona del crecimiento después de la pubertad.
P: ¿Qué causa el gigantismo?
R: El gigantismo está causado por una cantidad excesiva de hormona del crecimiento producida por la hipófisis antes de la pubertad.
P: ¿Cuál es la causa más común de la acromegalia?
R: La causa más común de la acromegalia es la presencia de un adenoma hipofisario, un tumor en la glándula pituitaria.
P: ¿Quién corre más riesgo de desarrollar acromegalia?
R: La acromegalia se diagnostica con mayor frecuencia en adultos de mediana edad.
P: ¿Cuáles son las posibles consecuencias de una acromegalia no tratada?
R: La acromegalia no tratada puede provocar desfiguraciones graves, complicaciones serias y la muerte prematura.
P: ¿Por qué es difícil diagnosticar la acromegalia?
R: La acromegalia es difícil de diagnosticar cuando acaba de empezar y no suele detectarse hasta 10-12 años después de su aparición.
P: ¿Cuáles son los síntomas visibles de la acromegalia?
R: El síntoma más notable de la acromegalia son los cambios en el aspecto físico de la persona, sobre todo en la cara.
Artículos relacionados
Autor
AlegsaOnline.com Acromegalia: qué es, causas, síntomas, diagnóstico y tratamiento Leandro Alegsa
URL: https://es.alegsaonline.com/art/762